
Translate this page into:
Phylo-geo haplotype network-based characterization of SARS-CoV-2 strains circulating in India (2020-2022)
For correspondence: Dr Sarah Cherian, Bioinformatics Group, ICMR-National Institute of Virology, Pune 411 001, Maharashtra, India e-mail: sarahcherian100@gmail.com
-
Received: ,
Abstract
Background & objectives
Genetic analysis of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) strains circulating in India during 2020-2022 was carried out to understand the evolution of potentially expanding and divergent clades.
Methods
SARS-CoV-2 sequences (n=612) randomly selected from among the sequences of samples collected through a nationwide network of Virus Research Diagnostic Laboratories during 2020 (n=1532) and Indian sequences available in Global Initiative on Sharing All Influenza Data during March 2020-March 2022 (n=53077), were analyzed using the phylo-geo haplotype network approach with reference to the Wuhan prototype sequence.
Results
On haplotype analysis, 420 haplotypes were revealed from 643 segregating sites among the sequences. Haplotype sharing was noted among the strains from different geographical regions. Nevertheless, the genetic distance among the viral haplotypes from different clades could differentiate the strains into distinct haplo groups regarding variant emergence.
Interpretation & conclusions
The haplotype analysis revealed that the G and GR clades were co-evolved and an epicentrefor the evolution of the GH, GK and GRA clades. GH was more frequently identified in northern parts of India than in other parts, whereas GK was detected less in north India than in other parts. Thus, the network analysis facilitated a detailed illustration of the pathways of evolution and circulation of SARS-CoV-2 variants.
Keywords
Evolutionary pathways
haplotype network
mutation analysis
phylogenetic analysis
population genetics
SARS-CoV-2

Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), the causative agent of the recent pandemic, belongs to the family Coronaviridae and genus Betacoronavirus. The virus is enveloped, large (80 to 120 nm in diameter), roughly spherical with unique surface projections, contains a single-stranded and positive-sense ribonucleic acid (RNA) genome (⁓29.8 kb in length) and transmits through respiratory route. Majority of SARS-CoV-2 infected patients manifest mild to moderate symptoms, like fever and respiratory symptoms, but a few may manifest severe acute respiratory distress syndrome (SARS)1,2.
Genomic surveillance to identify variants of concern and understand their effect on transmission, immune response, and disease severity is crucial to controlling the pandemic. Different variants are described by Global Initiative on Sharing All Influenza Data (GISAID) and Next-strain using a phylogenetic approach, while Pangolin is annotating them using a decision tree approach3-5.
Rapid sequencing, low variability and sequencing error have been challenging the direct use of phylogenetic methods on the SARS-CoV-2 genome alignments. Further, constructing reliable phylogenies from giant data sets, limited phylogenetically informative sites and weak phylogenetic signals due to sequencing errors and random mutations are confounding true evolutionary relationships, sometimes leading to monophyletic groups that are falsely resolved6,7. Hence, it is difficult, especially in large data sets of sequences, to understand the relation between different variants of concern and identify the genomic background from which they have emerged; however, understanding the history of occurrence of a specific variant is necessary for predicting its evolution and potential impact. Population genetic algorithms-haplotype networks have been commonly used in biology to characterize the evolution of genetic variations within closely related large datasets of sequences; however, they are less commonly used in virology8.
This study aimed to characterize SARS-CoV-2 strains circulating in India from March 2020 to March 2022 using a haplotype network approach. The overall purpose was to help understand the circulation of SARS-CoV-2 strains in different geographic regions of India and the evolution of potentially expanding and divergent clades.
Material & Methods
This study was undertaken at National Influenza Centre and Bioinformatics Group, ICMR-National Institute of Virology (ICMR-NIV), Pune from 2020-2022 after obtaining approval from Institutional Ethics Committee.
Sample collection and processing
Naso/oropharyngeal swab samples from suspected cases of Coronavirus disease 2019 (COVID-19) that were collected through Virus Research and Diagnostic Laboratories (VRDLs) in different regions of India (Supplementary Material I) and tested positive for SARS-CoV-2 through RT-PCR (by amplification of E, ORF, RDRP and ACTIN genes), were transferred to the nodal laboratory, ICMR-NIV, Pune, in cold chain for further processing. The samples stored appropriately at -80° C and with a cycle threshold of <30 were included in this study. Whole genome sequencing of SARS-CoV-2 in the naso/oropharyngeal samples (n=1532) was carried out.
Whole genome sequencing
Briefly, the extraction of viral RNA was carried out by viral RNA extraction kit (Qiagen, Hilden co., Germany) and was quantified using Qubit® 4.0 Fluorometer (Thermo Fisher Scientific, MA, USA). The RNA was reverse transcribed to complementary deoxyribonucleic acid (cDNA) using the SuperScript™ VILO™ cDNA Synthesis Kit (Invitrogen, CA, USA). Further, panel libraries were made using Ion AmpliSeq™ Library Kit Plus (Invitrogen, CA, USA). Template preparation was carried out using the Ion Chef System. The purified template beads were put forward for sequencing in the Ion S5 plus platform using the Ion 540™ chip as per the manufacturer’s instructions (ThermoFisher Scientific, MA, USA). The raw sequence data was processed by Torrent Suite Software (TSS) v5.10.1 (Thermo Fisher Scientific, MA, USA). Reads were assembled based on the Wuhan prototype reference sequence (Accession No.: NC_045512.2) using SARS-CoV2 plugins: variant Caller, Iterative Refinement Meta-Assembler9 and coverage analysis. The median coverage of the sequences of this study is found to be 95.96% at 20X depth. The detailed steps for data processing are described in Supplementary Figure 1. All the sequences are submitted to the public domain and are available at https://www.gisaid.org. The accession numbers are listed in Supplementary Material I.
Phylo-geo network analysis
For analysis of SARS-CoV-2 sequences circulating in different geographical regions of India (Supplementary Material I) during 2020-22 using a phylo-geo haplotype network approach, 612 SARS-CoV-2 complete genome sequences (>28000 bp in length) were randomly selected (by generating random numbers using MS Excel) from sequences obtained from samples that were collected through the nation-wide VRDL network during 2020 (n=1532) and Indian sequences available in GISAID from March 2020 to March 2022 (n=53077), along with the Wuhan prototype sequence (NC_045512). The dataset thus generated was ensured to be spatially and temporally representative. The list of sequences taken for this analysis is mentioned in supplementary Material I. The complete genome sequences were aligned by MAFFT v.7.4510 using default parameters. Variable sites in the multiple sequence alignment were selected using MEGA 6 software11. The viral haplotypes were identified from the variable sites using DnaSP v.6 software12. The network of the viral haplotypes was constructed by PopART v.1.7 software13 using the median-joining method (Epsilon=0); analysis of molecular variance: nucleotide diversity, fixation index/PhiST (a measure of evolutionary genetic distance among all pairs of viral haplotypes), Tajima’s D (mean number of pairwise differences/segregating sites) and the viral haplotype/clade distribution in map were also analyzed using the software. The maximum likelihood phylogenetic tree with 1000 bootstrap replications was constructed using IQ-TREE14 and FigTree v.1.4.415 was used for the tree visualization and annotation.
Results & Discussion
A median-joining haplotype network was constructed in comparison with phylogenetic trees to study the evolution of SARS-CoV-2 strains circulating in different geographical regions of India during 2020-2022. This study’s analysis of SARS-CoV-2 genomic sequences (n=612) revealed 171 parsimony informative (PI) sites among the 643 segregating sites. Nucleotide diversity among the sequences was noted as 0.0133. The negative value (-2.72) of statistic Tajima’s D denoted that these PI sites were significant in the evolution of these viral genomes. Allocation of each genome sequence to a haplotype was carried out through the detection of the mutation motif, which was haplotype-specific, based on the sites of segregation among the sequences, and that had revealed 420 different haplotypes (Supplementary Material II). The median-joining networks of the haplotypes (with traits coloured based on geographical region as in Supplementary Fig. 2 and clades as in Supplementary Fig. 3) were constructed, and the network topology (Supplementary Fig. 2 and 3) showed multiple star-like appearances, denoting the expansion of highly differentiated haplotypes. On analysis of molecular variance, the genetic distance found among the viral haplotypes from different geographical regions was insignificant (5.14%, PhiST=0.0514). Thus, the viral haplotypes of different geographical areas of this study showed a lot of haplotype sharing (Supplementary Fig. 2), which might indicate frequent travel inhuman populations among the geographical regions of this study. Due to human travel, previous investigators reported the distribution of haplotypes across geographical areas in the early phase of the pandemic in 20208,16-18.
Further, the analysis of molecular variance denoted a significant genetic distance among the viral haplotypes from different clades (60.07%, PhiST=0.6007), which differentiated the strains into distinct haplo-groups that showed a minimal haplotype sharing (Supplementary Fig. 3). Haplotypes viz., 9, 45, 107, 83, 160, 14 and 14 were identified among the haplogroups/clades: O, G, GH, GR, GK, GRA and others (S, GRY/alpha variant and GV), respectively. The circulation of the strains from the different clades in India during March 2020 – March 2022 are depicted in Figure. A few SARS-CoV-2 sequences of O and S clades in India that circulated during 2020, similar to the Wuhan prototype strain, were noted to have evolved into G and GR/gamma variant clades that circulated during 2020-21and were followed by subsequent evolutions into clades GH/beta variant that also circulated during 2020-21, GK/delta variant, that circulated during 2021 and GRA/ omicron variant that circulated during 2022 (Supplementary Fig. 3). Strikingly, G and GR clades co-evolved and became epicentres for the evolution of other clades. The GK that caused a wave in the pandemic during 2021, the GRA that caused a wave in the pandemic during 2022 and the viral strains from other less prevalent clades in India, such as GRY/alpha variant and GV clades, were found to arise from G-GR background only. Thus, the haplotype analysis concerning clades could throw light on the likely clade diversification pattern, emphasizing G-GR as the epicentre for the evolution of the other clades.
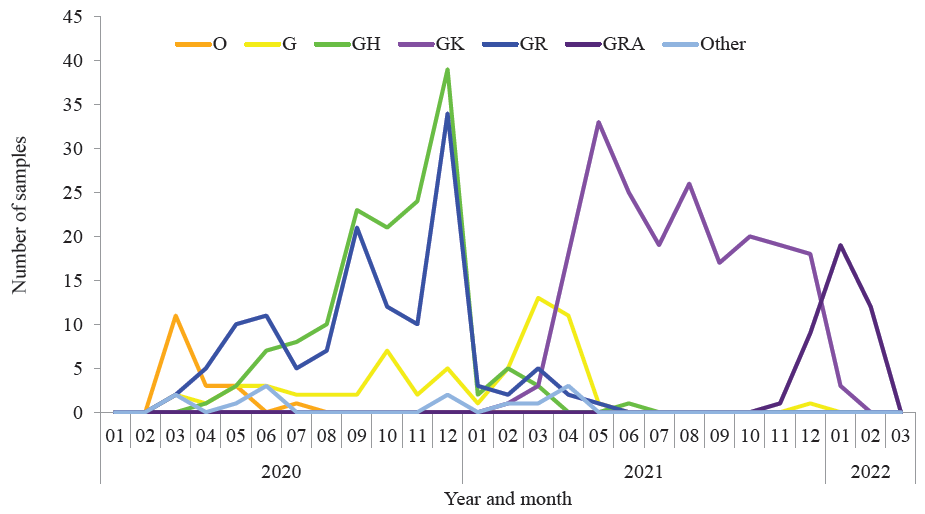
- . Circulation of the SARS-CoV-2 strains from different clades in India during March 2020-2022.
Pango lineages B.1, B.1.1, B.1.36, B.617.2 and BA.2 were predominantly found among the strains of G, GR, GH, GK and GRA clades, respectively, while B.1 and B.1.1.7 lineages represented co-evolution/emergence of G, GR and GH clades (Supplementary Fig. 4). Mutation analysis with respect to the Wuhan prototype reference sequence showed that the amino acid substitutions D614G in spike and P323L in NSP12_RdRp proteins, which were earlier found to be associated with the predominantly transmitting strains19, were frequently detected in strains across the G, GH, GR, GK and GRA clades in this study (Supplementary Fig. 5). These findings were in agreement with that of other investigators20,21.
Analysis of the distribution of clades among different geographical regions of India had denoted that strains from clade GH reported predominantly during the first wave in 2020 were more frequently identified among the genomes submitted from northern parts of India than those from other parts of India, whereas the strains from clade GK reported predominantly during the second wave in 2021 were detected less in northern India than that in other parts (Supplementary Fig. 6). Notably, the clade distribution (Supplementary Fig. 6) showed the prevalence of an average of three clades per region. Besides the study sample set, this scenario was reflected among all the sequences submitted to GISAID from March 2020 to March 2022 (data not shown). Similarly, distribution bias of different clades among geographical regions has been noted during 2020 by other investigators22,23. However, further studies would be essential to understand the factors affecting the distribution bias of different SARS-CoV-2 clades. An evolutionary stasis was noted relatively in 2020 during the initial phase of the pandemic, while variants of concern such as delta and omicron that acquired selective advantages to the host immune system were found to be emerging over time due to mutational jumps (Figure; Supplementary Fig. 3, 4 and 5). This observation agrees with a few previous studies from other countries24-27.
The maximum likelihood phylogenetic trees were constructed using individual viral sequences (Supplementary Fig. 7) and viral haplotype sequences (Supplementary Fig. 8) to compare the above results with reference to the direct use of phylogenetic methods. The value addition in the case of the haplotype network (Supplementary Fig. 3) in terms of emerging lineages/pathways of the virus’s evolution through the topology was evident when compared with the phylogenic trees constructed based on either the individual viral sequences (Supplementary Fig. 7) or viral haplotype sequences (Supplementary Fig. 8). Similar observations were reported in other studies as well6-8.
By using the haplotype network approach that uses the most frequent mutational events and viral haplotypes with common variants, we could note a more explicit inference and clarification of the genetic background of variants of concern, better visualization of the pathways of evolution among different variants and efficient identification of recurrent mutations and steps in the emergence of the variants.
In conclusion, this study characterized the SARS-CoV2 strains circulating in India during 2020-2022. The findings indicated G-GR as the epicentre for the evolution of several other clades of SARS-CoV2 and differential circulation of GH and GK clades in different geographical regions of India during March 2020–March 2022. The study also reiterated that haplotype network approaches were effective and more robust than the surveillance strategies based only on phylogenetic trees, especially in large-scale analysis of less variable viral populations such as SARS-CoV-2. In the recent SARS-CoV-2 pandemic scenario, understanding the viral evolutionary strategies was crucial to identify emerging variants that might fit better as vaccine-escapes or more virulence. The knowledge generated by us might be helpful in designing efficient preventive and therapeutic strategies during possible resurgence, if any, in future.
Declaration
Supplementary Figure 3 was previously published in the Annual Research Progress Report, ICMR-National Institute of Virology, Pune (2021-2022) (https://niv.icmr.org.in/images/pdf/annual_reports/Annual_Report_2021_2022.pdf).
Financial Support & Sponsorship
The authors thank ICMR-NIV Pune for all the financial support to this study and DST for WOS-B fellowship (DST order no. DST/WOS-B/ER-1/2021) to the second author ‘RL’.
Conflicts of Interest
None.
Use of Artificial Intelligence (AI)-Assisted Technology for manuscript preparation
The authors confirm that there was no use of Artificial Intelligence (AI)-Assisted Technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 2021;19:155-70.
- [Google Scholar]
- A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med. 2020;382:727-33.
- [Google Scholar]
- Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob Chall. 2017;1:33-46.
- [Google Scholar]
- Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021;7:veab064.
- [Google Scholar]
- Next strain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34:4121-3.
- [Google Scholar]
- Phylogenetic Analysis of SARS-CoV-2 Data Is Difficult. Mol Biol Evol. 2021;38:1777-91.
- [Google Scholar]
- Bioinformatics resources for SARS-CoV-2 discovery and surveillance. Brief Bioinform. 2021;22:631-41.
- [Google Scholar]
- Phylogenetic network analysis of SARS-CoV-2 genomes. Proc Natl Acad Sci U S A. 2020;117:9241-3.
- [Google Scholar]
- Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler. BMC Genomics. 2016;17:708.
- [Google Scholar]
- MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059-66.
- [Google Scholar]
- MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30(12):2725-9.
- [Google Scholar]
- DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol Biol Evol. 2017;34:3299-302.
- [Google Scholar]
- PopART: full-feature software for haplotype network construction. Methods Ecol Evol. 2015;6:1110-16.
- [Google Scholar]
- UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol Biol Evol. 2018;35:518-22.
- [Google Scholar]
- Available from: http://tree.bio.ed.ac.uk/software/figtree, accessed on February 2, 2023.
- Phylo-geo-network and haplogroup analysis of 611 novel coronavirus (SARS-CoV-2) genomes from India. Life Sci Alliance. 2021;4:e202000925.
- [Google Scholar]
- In silico comparative genomics of SARS-CoV-2 to determine the source and diversity of the pathogen in Bangladesh. PLoS One. 2021;16:e0245584.
- [Google Scholar]
- Spike protein D614G and RdRp P323L: the SARS-CoV-2 mutations associated with severity of COVID-19. Genomics Inform. 2020;18:e44.
- [Google Scholar]
- Genomic characterization and epidemiology of an emerging SARS-CoV-2 variant in Delhi, India. Science. 2021;374:995-9.
- [Google Scholar]
- SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature. 2021;599:114-9.
- [Google Scholar]
- Global dynamics of SARS-CoV-2 clades and their relation to COVID-19 epidemiology. Sci Rep. 2021;11:8435.
- [Google Scholar]
- An Epidemiological Analysis of SARS-CoV-2 Genomic Sequences from Different Regions of India. Viruses. 2021;13:925.
- [Google Scholar]
- Ongoing global and regional adaptive evolution of SARS-CoV-2. Proc Natl Acad Sci USA. 2021;118:e2104241118.
- [Google Scholar]
- SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. 2021;19:409-24.
- [Google Scholar]
- Population Genomics Approaches for Genetic Characterization of SARS-CoV-2 Lineages. Front Med (Lausanne). 2022;9:826746.
- [Google Scholar]




