Translate this page into:
Temporal cytokine expression and the target organ attributes unravel novel aspects of autoimmune arthritis
Reprint requests: Dr Kamal D. Moudgil, Professor, Department of Microbiology & Immunology, University of Maryland School of Medicine, 685 W. Baltimore Street, HSF-1, Suite 380, Baltimore, MD 21201, USA e-mail: kmoud001@umaryland.edu
-
Received: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Susceptibility to autoimmunity is determined by multiple factors. Defining the contribution of the quantitative versus qualitative aspects of antigen-directed immune responses as well as the factors influencing target organ susceptibility is vital to advancing the understanding of the pathogenesis of autoimmunity. In a series of studies, we have addressed these issues using the adjuvant-induced arthritis (AA) model of human rheumatoid arthritis (RA). Lewis rats are susceptible to AA following immunization with heat-killed Mycobacterium tuberculosis H37Ra, whereas Wistar-Kyoto (WKY) rats of the same MHC (major histocompatibility complex) haplotype are resistant. Comparative studies on these and other susceptible/resistant rodent strains have offered interesting insights into differential cytokine responses in the face of comparable T cell proliferative response to the disease relevant antigens. Study of the cytokine kinetics have also permitted validation of the disease-protective versus disease-aggravating effects of specific cytokines by treatment of rats/mice with those cytokines at different phases of the disease. In regard to the target organ attributes, the migration of arthritogenic leukocytes into the joints; the expression of mediators of inflammation, angiogenesis, and tissue damage; the role of vascular permeability; and the characteristics of vascular endothelial cells have been examined. Further, various inhibitors of angiogenesis are effective in suppressing arthritis. Taken together, the differential cytokine responses and unique attributes of the target organ have revealed novel aspects of disease susceptibility and joint damage in AA. The translation of this basic research in animal models to RA patients would not only advance our understanding of the disease process, but also offer novel avenues for immunomodulation of this disease.
Keywords
Adjuvant arthritis
angiogenesis
arthritis
autoimmunity
cytokines
inflammation
joints
matrix metalloproteinases
regulatory T cells
T helper cells

Introduction
The immune system is capable of effectively responding to and containing a wide variety of pathogens (foreign; non-self), while guarding against immune response to the host tissues (self)123. However, certain constellations of genetic and environmental factors may result in a breakdown of self tolerance resulting in anti-self immune response (autoreactivity), which if not regulated, can result in immune pathology and dysfunction (autoimmunity). At the cellular level, the induction of autoimmunity is a manifestation of an imbalance between pathogenic effector versus protective regulatory responses. The manifestations of autoimmunity may either be systemic or organ-specific. The traditional view of this dichotomy is based on the distribution of the antigen targeted by the autoimmune response, with widely-distributed autoantigens invoked in systemic diseases, while tissue-restricted self antigens implicated in organ-specific diseases. Although this scheme can explain the distribution of immune pathology in the body in many diseases, but it fails in other situations. The latter instances are those where the autoimmune response is directed against ubiquitously distributed antigens, yet the primary target of autoimmune damage may be limited to a particular tissue/organ45.
It is increasingly being realized that the target antigen in organ-specific immunity may not necessarily be unique to that particular tissue/organ. Instead it may be distributed widely and yet the immune pathology may be predominantly focused on one or a few organs. This is illustrated in a couple of animal models of autoimmune arthritis. Adjuvant-induced arthritis (AA) in the rat is a well-studied model of human rheumatoid arthritis (RA)67. It can be induced in the Lewis rat by immunization with heat-killed Mycobacterium tuberculosis H37Ra (Mtb). AA is a T cell-mediated disease. Interestingly, immune response against mycobacterial heat-shock protein 65 (Bhsp65) has been implicated in the immunopathogenesis of AA5891011121314. Given the highly conserved nature of heat-shock proteins (Hsps), the T cells and antibodies directed against Bhsp65 are crossreactive with self hsp65 or other self ligands that mimic the foreign hsp65 epitopes. Further, Mtb also contains other heat-shock proteins besides Bhsp65. Hsp65 and other members of the Hsp60 family have been invoked not only in arthritis but also in multiple sclerosis (MS) and type I diabetes mellitus (T1D)8151617. However, Mtb-immunized Lewis rats develop arthritis without any concurrent autoimmune damage to the central nervous system or the pancreatic β-islet cells. The latter two represent the target organs in MS and T1D, respectively and their corresponding animal models are experimental autoimmune encephalomyelitis and the non-obese diabetic mice.
Another example of the animal model of arthritis in which the autoimmune response is directed against a ubiquitously distributed antigen is the K/BxN model of arthritis418. In this model, mice bearing a transgenic T cell receptor (TCR) specific for an epitope within ribonuclease, when crossed with non-obese diabetic (NOD) mice, develop spontaneous arthritis18. Interestingly, the above-mentioned TCR fortuitously crossreacts with a glycolytic enzyme, glucose 6-phosphate isomerase (GPI). Thus, spontaneous arthritis in these mice is the result of an autoimmune response against GPI, a widely distributed antigen.
The above examples relating to arthritis and similar ones involving other autoimmune diseases have given credence to the idea that the target organ attributes might play a vital role in their susceptibility to autoimmunity over and above the basic preconditions for the breakdown of self tolerance and the induction of autoreactivity. Broadly, the factors influencing the target organ susceptibility can be grouped into those that are extrinsic to that organ and others that are intrinsic. Extrinsic factors include, for example, the quantitative and qualitative aspects of the immune response generated in the peripheral lymphoid tissue draining the site of antigenic challenge or antigen encounter12192021, and the kinetics of proinflammatory versus anti-inflammatory cytokines during the course of autoimmune arthritis2223. Intrinsic factors include the angiogenic process associated with arthritis2425, the local vasculature and its permeability4, the characteristics of the vascular endothelium of the joints26, and the local release of immunological and biochemical mediators of tissue damage27282930. This article addresses specific examples of both extrinsic and intrinsic factors involved in the target organ damage in autoimmune arthritis. Most of the description is based on the rat AA model. However, at several places, examples from other animal models of arthritis have also been discussed. Further, some basic information has also been included on the subsets of T helper and regulatory T cells, the key pro-inflammatory cytokines, the inducers and regulators of angiogenesis, and the matrix metalloproteinases. All these cellular/soluble mediators play critical roles in the disease process in arthritis.
Subsets of T helper cells and regulatory T cells involved in the pathogenesis of autoimmunity
T helper cells: The majority of animal models of RA are T cell-mediated diseases29 similar to the human disease31. The CD4+ helper T cells play an important role in the initiation and progression of acute inflammation in situations involving immune response to self (autoimmunity) or foreign (e.g. infectious disease) antigens. The cytokines produced by these cells help to facilitate the activation and chemotactic migration of other cell types to the site of inflammation during the immune response. The most widely studied helper CD4+ T cell types are the T helper 1 (Th1), Th2, and Th17. The cytokines produced by dendritic cells (DC) during an immune response are among the important factors that dictate the differentiation of T helper progenitor cells into specific subsets32 (Fig. 1). For example, when activated DCs and macrophages produce interleukin (IL)-12, it activates the ‘signal transducer and activator of transcription’ (STAT) 4 pathway leading to the upregulation of the transcription factor ‘T-box expressed in T cells’ (T-bet). T-bet induces Th1 cell differentiation resulting in upregulation of interferon (IFN)γ and downregulation of ‘GATA binding protein 3’ (GATA3) and IL-43233. The Th1 cell produces IFNγ and promotes cellular immune response. In contrast, IL-4 signaling through STAT6 leads to a Th2 differentiation, upregulation of GATA3, and downregulation of IFNγ34.
The Th2 cells produce IL-4, IL-5, and IL-13 and lead to B cell proliferation and antibody production. In this scheme of the two polar types of Th cells, Th1 and Th2, the immune responses and the diseases associated with them could be categorized as predominantly Th1- or Th2-mediated diseases. With the ability of Th1 to regulate Th2, and vice versa, the Th1-Th2 paradigm provided a fine conceptual framework to comprehend immune responses during health and disease. However, this Th1-Th2 paradigm had to be expanded and revised with the discovery of IL-23, which shares the p40 subunit with IL-12, and thereby was responsible for many of the immune effects that had previously been attributed to IL-12, or the lack of it35. In addition, newer cytokines (e.g. IL-17, IL-21, and IL-22) associated with inflammation were reported, which helped envision additional families of Th cells such as Th17 and Th2229. It has now been shown that Th17 cells are involved in many autoimmune diseases once thought to be primarily Th1-driven, including RA36. The Th17 cells produce IL-17, IL-21, IL-22, and IL-23, and play a role in RA and mucosal immunity37. In mice, the Th17 cells differentiate from naïve T cells in the presence of transforming growth factor (TGF) β and IL-6. This process involves signaling through STAT3 and increased expression of the transcription factor ‘retinoic acid receptor-related orphan receptor gamma t’ (RORγt)38. Alternatively, Th17 cells can differentiate in the presence of IL-21 and TGFβ, as in the case of deficiency of IL-6. However, in the absence of IL-6, T cells preferentially differentiate into Forkhead box p 3 (Foxp3)-expressing CD4+CD25+ Foxp3+ TReg cells, indicating that IL-6 is a regulator of the balance between Th17- TReg39.
Regulatory T cells: There are several types of regulatory T cells. One of the recent additions to this group is the TReg cell. The phenotype of the TReg is CD4+CD25+Foxp3+. These cells can either differentiate naturally in the thymus (nTReg) or be induced in the periphery (iTReg). The differentiation of TReg requires TGFβ40. TReg cells suppress effector cells by producing TGFβ and IL-10 and via cytotoxic T-lymphocyte antigen (CTLA)-4 expressed on their cell surface. A subset of CD8+CD25+ Foxp3+ TReg has also been described41. Other regulatory subsets are Tr 1 and Th 342. The Tr 1 cell is a CD4+CD25+Foxp3- T cell that requires IL-10 for its differentiation, and it secrets IL-10. The Th3 cell is a regulatory cell associated with the gut mucosa. It was shown to mediate the immunosuppressive effects of tolerance to the antigens administered via oral route (oral tolerance). Th3 cells mediate their suppressive action via secretion of TGFβ42. The balance between the effector T cells and the regulatory T cells determines whether or not autoreactive cells can induce an autoimmune response.
The effector functions of the cytokines that play a key role in arthritis pathogenesis
In RA and animal models of arthritis, inflammatory cytokines play a pivotal role in driving the disease process (Table I). Accordingly, biologic medications targeting these cytokines are being used for the treatment of arthritis. The cytokines that have been studied extensively are TNFα, IL-1, and IL-643. TNFα has been at the center of RA research for several years because of its vital role in joint destruction and the control over other proinflammatory cytokines. It has been shown that TNFα controls the production of IL-1β and IL-8 by synovial cells46. In addition to increasing the release of other cytokines, TNFα can increase cellular infiltration into the synovium by enhancing chemokine expression, endothelial cell activation, and angiogenesis43. Finally, TNFα and IL-1 cause bone damage, the hallmark of RA pathogenesis4748. In animal models of arthritis, IL-1β works in conjunction with IL-6 during the early phases of the disease, acting on endothelial cells to secrete chemokines like IL-8 and monocyte chemotactic protein (MCP)-1 to attract monocytes. IL-6 further upregulates chemokines that attract T cells, leading to enhanced cellular infiltration and beginning the transition from an acute inflammatory disease to a chronic immune disease49. These two phases are dominated by innate and adaptive immunity, respectively.
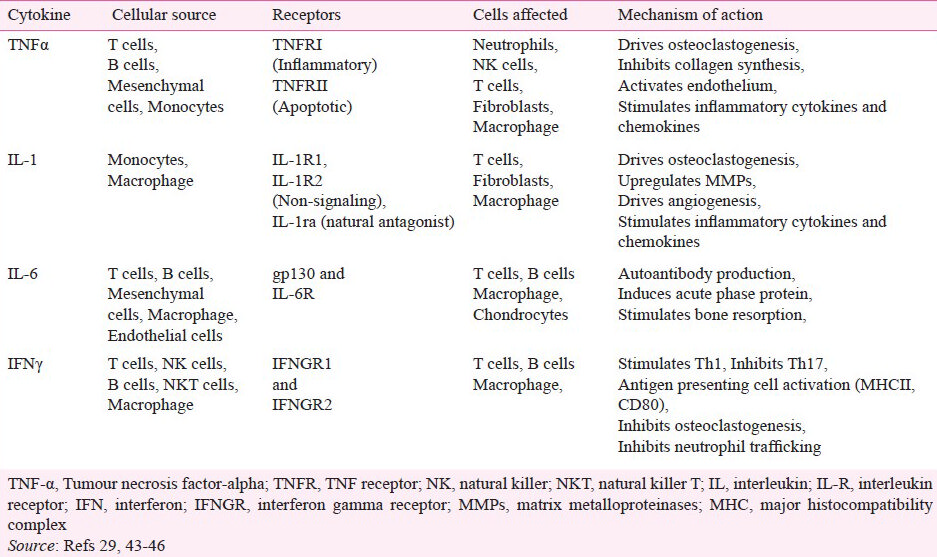
IL-17 is involved in the inflammatory response and it has been implicated in the pathogenesis of several autoimmune diseases including RA50 and multiple sclerosis51. IL-17 is predominantly produced by the CD4+ Th17 cells. Other sources of IL-17 include CD8+ T cells, γδ T cells, natural killer T (NKT) cells, and lymphoid tissue inducer (LTi) cells5253. There are six isoforms of IL-17, with IL-17A most commonly referred to as IL-17. IL-17 is a proinflammatory cytokine that plays an important role in inflammation of the synovium during RA. IL-17 can help promote the production of inflammatory cytokines like IL-6 and leukemia inhibitory factor (LIF)50, and matrix degrading enzymes, matrix metalloproteinase (MMP) 1 and 3 by synovial fibroblasts54. IL-17 also induces osteoclastogenesis by upregulating receptor activator of NFκB (RANK) on osteoclast precursors and its ligand RANKL on the surface of activated T cells, resulting in bone erosion55. IL-17 also facilitates cellular infiltration either directly or by increasing the expression of chemokine (C-C motif) ligand (CCL) 20, CXC motif chemokine ligand (CXCL) 12, and CXCL5, and attracting B cells, T cells, neutrophils, and monocytes to the synovium565758. The pannus formation (cellular infiltration into hyperplastic vascularized synovium) in an arthritic joint is enhanced further by the induction of IL-17-induced angiogenesis59.
IL-17 can be regulated by other cytokines. IL-1β, TNFα, and IL-23 increase IL-17 expression29353637, while IFNγ decreases IL-17 expression in autoimmune arthritis models27. IFNγ has for years been invoked as one of the major proinflammatory cytokines contributing to the pathogenesis of autoimmune diseases until the discovery of IL-17. However, recent studies have unraveled an opposite, anti-inflammatory role of IFNγ in arthritis models27. IFNγ-deficient mice showed an exacerbation of autoimmune arthritis and an increase in IL-1760. Similarly, we observed in the adjuvant arthritis model that IFNγ was expressed at the highest levels in the recovery phase of the disease22. Further, the treatment of rats either during the incubation phase or after disease onset with exogenous IFNγ reduced disease severity27. In addition, pre-treatment of rats with a C-terminal epitope 465-479 of rat heat-shock protein 65 (Rhsp65) suppressed arthritis, increased IFNγ production, but reduced IL-17 expression22.
IFNγ functions as a regulatory cytokine, directly as well as indirectly, in autoimmune arthritis. One indirect effect that IFNγ has is the upregulation of IL-27, a cytokine that can reduce IL-17 production. IL-27 is an IL-12 superfamily cytokine, and it can prevent the differentiation of Th17 by reducing both the expression of RORγt and the phosphorylation of STAT3. Both of these transcription factors are integral to the differentiation of Th1727. IL-27 is comprised of two subunits, Epstein-Barr virus-induced gene 3 (EBI3) and p28. It is secreted by macrophages, dendritic cells, epithelial cells and a wide range of innate and adaptive immune cells, most notably the CD4+ T cells61. In addition to preventing pathogenic T cell differentiation, IL-27 can induce both the expression of program death-ligand 1 (PD-L1), a co-receptor that is a negative regulator of T cell function56 , and the generation of IL-10-producing Tr1 cells62. IL-27 is also able to downregulate the expression of RANK on osteoclast precursors and RANKL on CD14+ cells, causing a decrease in osteoclastogenesis and thereby limiting bone loss63. Though the role of IL-27 in autoimmunity has not yet been fully defined, increasing evidence points to its anti-inflammatory and anti-arthritic activities.
Kinetics of cytokine expression during the course of arthritis and its correlation with disease susceptibility versus resistance
Animal models of human RA29 permit comprehensive experimental studies on the pathogenesis of autoimmunity, including the role of T cells and cytokines in the disease process. Two of the commonly used models of RA are adjuvant arthritis (AA) in rats7 and collagen-induced arthritis (CIA) in mice/rats64. The cytokines released during the course of autoimmune arthritis influence the severity of the pathological and clinical features of the disease (Figs 1 and 2). The AA model system has extensively been used to examine the disease-related events at different time points in the course of the disease as well as for testing potential anti-arthritic agents for their therapeutic efficacy and side effects. However, not all rat strains are equally susceptible to autoimmune arthritis. The Lewis rat is highly susceptible to AA, whereas the Wistar-Kyoto (WKY) rat is resistant to arthritis despite having a similar major histocompatibility complex (MHC) haplotype as the Lewis rat51121. WKY rats provide a good control for Lewis rats for studies on disease pathogenesis. In our studies, we have exploited this pair of rat strains for addressing important aspects of AA, for example, epitope mapping of the disease-related antigen Bhsp65511 , cellular migration into the joints67 , the dynamics and epitope reactivity of antibodies produced during AA68 , and cytokine responses against Bhsp6522232769.
In different studies performed in the AA model, cytokine responses have been examined in the draining lymph nodes, spleen, synovial-infiltrating cells (SIC), or joint homogenates. Also, not all time points have been tested in each tissue. This makes it somewhat difficult to directly compare the profiles obtained using one tissue with that derived from another tissue. However, it is understandable that the peripheral lymphoid tissues and SIC might show similarities in certain cytokine responses, but differences in others. A set of representative profiles of pro-/anti-inflammatory cytokines in AA constructed from the results of different studies2223276566 is shown in Fig. 2. In rats with AA, the expression of pro-inflammatory cytokines (TNFα, IL-1 and IL-6) showed a gradual increase from the incubation phase to the ‘onset’ phase. After that point, IL-1 and IL-6, but not TNFα levels decreased during the regression phase. Surprisingly, IL-17 expression was highest in the incubation phase of the disease, but thereafter its levels remained low throughout the disease. In contrast, the expression of IFNγ, IL-27, and IL-10 was low during the early incubation and onset phases of the disease, but it increased during the late stages correlating with disease regression. IL-10 is a well known anti-inflammatory cytokine. Our results point towards a disease-regulating role of IFNγ and IL-27 in AA. IFNγ is considered to be a prototypic pro-inflammatory cytokine. However, our studies have shown that this cytokine also possesses arthritis-suppressive activity22. In the case of IL-27, which is among the newer cytokines whose functional attributes have not yet been fully defined, our results27 clearly demonstrate that it has anti-arthritic activity. For this reason, we have depicted IFNγ, IL-27, and IL-10 under anti-inflammatory cytokines (Fig. 2).
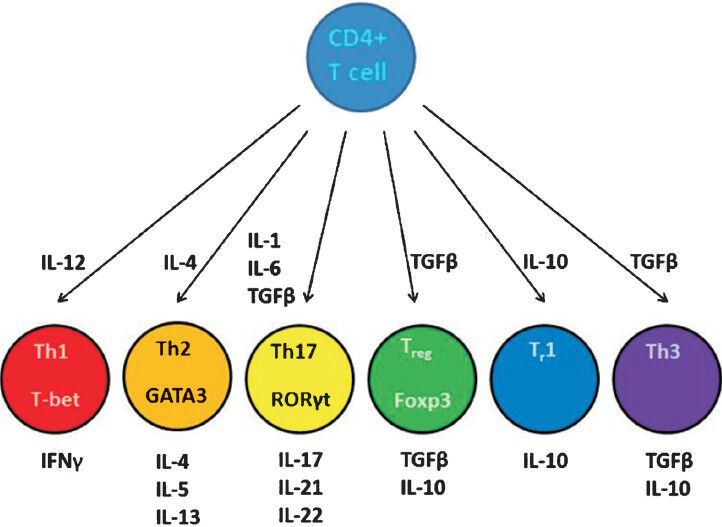
- CD4+ T cell differentiation and cellular phenotype. Naïve CD4+ T cells can differentiate into several different types of T helper (Th) or regulatory T (Treg or Tr) cells depending on the cytokine environment in which these are activated. The regulating transcription factors of these subsets and their characteristic effector cytokines are shown. (CD, cluster of differentiation; GATA3, GATA binding protein 3; T-bet, T-box expressed in T cells; RORγt, retinoic acid receptor-related orphan receptor gamma; Foxp3, Forkhead box p3; IL, interleukin; IFN, interferon; TGF, transforming growth factor.
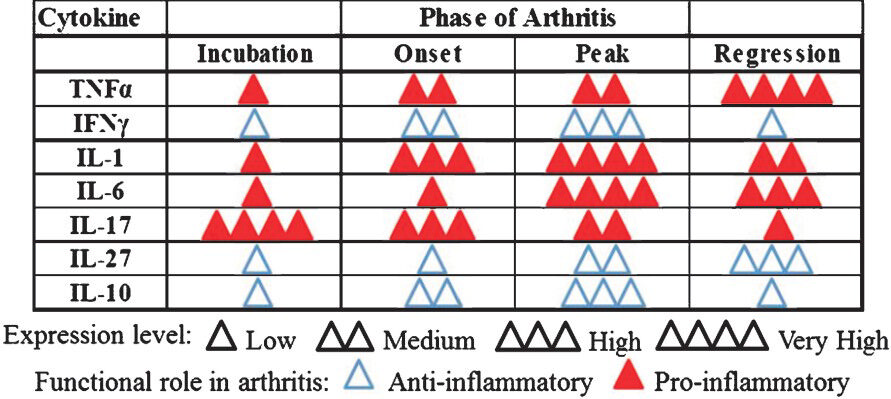
- The dynamics of cytokine expression during the course of adjuvant arthritis. Adjuvant arthritis (AA) in the Lewis rat, induced following immunization with heat-killed M. tuberculosis H37Ra, displays distinct phases of the disease. These phases include incubation, onset, peak and regression. Proinflammatory cytokines play a vital role in the initiation and progression of arthritis, whereas anti-inflammatory cytokines facilitate regression of inflammatory arthritis. The levels of cytokines represented by the number of triangles are relative to each phase for that particular cytokine. (IL, interleukin; IFN, interferon; TNF, tumour necrosis factor).
-
Source: Refs 22, 23, 27, 65, 66
In contrast to the AA-susceptible Lewis rats, the AA-resistant WKY rats had a different cytokine profile that helped us comprehend the lack of signs of disease following an arthritogenic challenge27. The pattern of expression of IL-17 in Wky0 rats was similar to that of Lewis rats. However, unlike Lewis rats, WKY rats also showed highest levels of expression of IL-27 and IFNγ at the same time as that of IL-17. We suggested that the concurrent expression of these two cytokines with IL-17 helped neutralize the pathological effects of IL-17 in WKY rats27. In contrast, IL-17 activity was unopposed in the incubation period in the case of Lewis rats, thereby explaining the development of arthritis.
Our results of modulation of AA by treatment of Lewis rats with exogenous cytokine yielded interesting results27. The treatment of Lewis rats with IL-17 in the incubation phase of the disease leads to disease aggravation as expected from the pathogenic role of IL-17 in AA. The most interesting finding was the effect of IL-27 treatment on the inhibition of AA. These results demonstrate that IL-27 plays an immunoregulatory role in AA27. Surprisingly, the treatment of rats with IFNγ or TNFα also offered protection against AA, showing that the timing of administration of certain cytokines that are typically considered to be proinflammatory (e.g. IFNγ and TNFα) may also lead to the suppression of arthritis222327. This emphasizes the dual role of these proinflammatory cytokines69.
Taking together, studies on the temporal expression of cytokines in the AA-susceptible/-resistant rats have provided important and useful information regarding arthritis development and its deliberate control by cytokine treatment. For example, the cytokine profiles have provided insights into the relative contribution of different cytokines in the initiation and progression of AA, followed by the regression of inflammation (Fig. 2). Further, comparative studies in Lewis/WKY and other AA-susceptible/resistant rodent strains have offered interesting insights into differential cytokine responses in the face of comparable T cell proliferative response to the disease-related antigens. In addition, such studies have underscored the significance of the balance between the pro- and anti-inflammatory cytokines in determining (in part) whether arthritic inflammation would result or not following an arthritogenic stimulus, such as Mtb injection. Finally, the study of the cytokine kinetics has also permitted validation of the disease-protective versus disease-aggravating effects of specific cytokines as tested by the treatment of rats/mice at different phases of the disease.
Cellular migration into the joints and characteristics of the synovial-infiltrating cells
Cellular infiltration into the synovium is a characteristic feature of RA. The resulting pannus invades the surrounding bone and cartilage and leads to tissue damage, pain and disability in the affected joints. There are various cell types that infiltrate the synovium of arthritic joints. The T cell migration into the synovium plays an important role in the activation and recruitment of other cell types. Most notably, Th17 cells that migrate into the synovium lead to neutrophil recruitment via IL-17-mediated induction of chemokines70. It has recently been shown that IL-17-induced neutrophil recruitment occurs indirectly through TNFα, which binds its receptor TNF receptor (TNFR)171. Treg cells have also been isolated from inflamed synovium71 , but at present the relative kinetics and frequencies of Th17 and Treg in the joints during the course of arthritis in animal models or RA patients are not yet defined.
Other cell types that are found in arthritic joints are macrophage- and fibroblast-synoviocytes, neutrophils, B cells, dendritic cells, and mast cells72. This cellular migration followed by accumulation of cells in the joints correlates with arthritis susceptibility as shown in our comparative study in the AA model using arthritis-susceptible Lewis rats and arthritis-resistant WKy0 rats67. Chemokines play an important role in cellular migration into the joints. Chemokines are upregulated by cytokines and can reciprocate, and lead to the increased cytokine expression causing persistent inflammation73. As a result, chemokines can be targeted directly or indirectly through the suppression of cytokines to treat RA. We have recently shown that a natural plant product (celastrol) can decrease inflammatory cytokine and chemokine production leading to the inhibition of cell migration and suppression of arthritis in the AA model74.
Angiogenesis and its role in the pathogenesis of arthritis
Angiogenesis refers to the formation of new blood vessels from existing vessels, and it facilitates the nourishment and maintenance of growing tissue. Angiogenesis is considered to be one of the key mechanisms that promote chronic joint inflammation in RA75. During RA, hyperplastic synovium with immune cell infiltrates forms the pannus, which requires supply of oxygen and nutrients. Angiogenesis contributes to the formation and maintenance of the pannus in RA. The pannus is highly vascularized and it invades the articular cartilage and bone76. The actions of cytokines and other mediators of inflammation produced by the synovial-infiltrating cells result in cartilage damage and bone erosion in arthritic joints7677. Inhibition of angiogenesis by interfering with pathways driven by the vascular endothelial growth factor (VEGF) or other mediators results in delayed onset and reduced severity of arthritis in animal models, such as CIA in mice and AA in rats7879808182. These observations provide additional support for the role of angiogenesis in the initiation and propagation of RA.
Angiogenesis involves multiple cell types and complex interplay of mediators and pathways (Table II). The formation of new vessels from a pre-existing vessel is a result of a series of events including selective degradation of vascular basement membrane and adjacent extracellular matrix, and migration of endothelial cells leading to tube formation and capillary growth9091. VEGF is a key factor mediating angiogenesis. The levels of VEGF in the serum and synovial fluid correlate with the severity of RA. High requirement of oxygen in the pannus, the growing tissue in arthritic joints, makes that tissue relatively hypoxic. Hypoxia induces gene expression of pro-angiogenic proteins such as VEGF via the activation of transcription factors known as hypoxia-inducible factors (HIFs)91. Activation of HIFs can also occur by proinflammatory cytokines. VEGF in turn recruits monocytes, which differentiate into macrophages and secrete matrix-degrading enzymes known as matrix metalloproteinases (MMPs). VEGF also induces the migration and proliferation of endothelial cells and smooth muscle cells, which lead to the formation of new capillaries788392. Epidermal growth factor (EGF), fibroblast growth factor-2 (FGF-2), hepatocyte growth factor (HGF), insulin-like growth factor (IGF), and platelet-derived growth factor (PDGF) serve as additional mediators of angiogenesis in RA259293 (Table II).
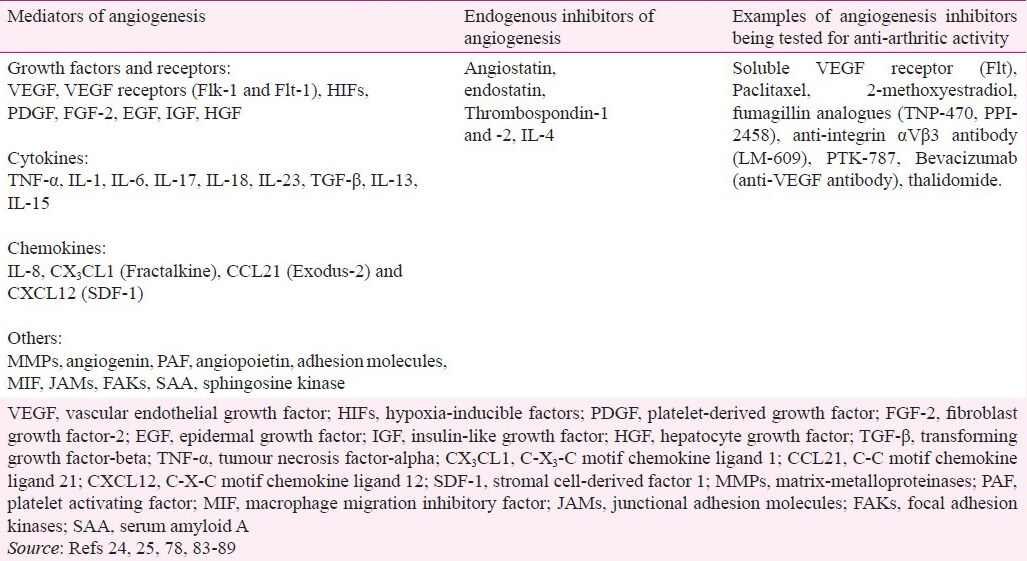
Several cytokines such as TGF-β, TNF-α, IL-1, IL-6, IL-8, IL-13, IL-15, and IL-18 modulate the process of angiogenesis84949596979899100101102 (Table III). For example, TGF-β and IL-1 stimulate the secretion of VEGF through the activation of HIF. These events are mediated via mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathway. The above-mentioned cytokines further enhance VEGF secretion induced by hypoxia. This cooperation between cytokines and hypoxia facilitates the secretion of large amounts of VEGF by synovial cells in the hypoxic environment in the arthritic joint resulting in increased angiogenesis.
A variety of chemokines contribute to the angiogenic process in RA (Table II). The expression of CXCL12 [or, stromal cell-derived factor 1 (SDF-1)], is influenced by cytokines and hypoxia. SDF-1 in turn facilitates the recruitment of lymphocytes into the arthritic synovium85. The chemokine CX3 CL1 (or, fractalkine), also induces angiogenesis103. CCL21 (Exodus-2) induces endothelial cell migration and tube formation. These events involve PI3K104.
Other mediators that influence angiogenesis include angiogenin, platelet activating factor (PAF), angiopoietin, soluble adhesion molecules, and endothelial mediator endoglin. Further, macrophage migration inhibitory factor (MIF), junctional adhesion molecules (JAMs) and focal adhesion kinases (FAKs) are associated with inflammatory angiogenesis. Serum amyloid A (SAA) and sphingosine kinase have also been invoked in the angiogenic events25 (Table II).
Matrix metalloproteinases (MMPs) and other cartilage-degrading enzymes
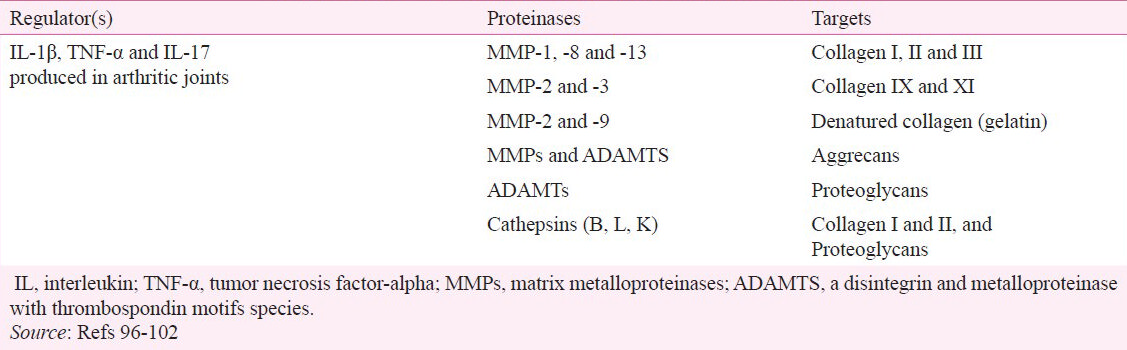
Articular cartilage, which is made of extracellular matrix (ECM) and chondrocytes, is the major tissue targeted in the joints in RA. In uncontrolled RA, there is a progressive damage to the joints leading to physical disabilities. Cartilage ECM contains primarily proteoglycans, aggrecan and collagen. Aggrecan associates with proteoglycans to form a network of collagen fibers, which are the main structural component of the matrix105. Articular cartilage is mainly composed of type II collagen, but it also contains collagen type IX XI and VI. The cartilage matrix also contains leucine-rich proteoglycans, including decorin, fibromodulin and biglycan105. Cartilage degradation can be mediated by a variety of proteases including MMPs, cathepsins (B, L, K) and “a disintegrin and metalloproteinase with thrombospondin motifs species” (ADAMTS)96979899100101102 (Table III).
Cartilage degradation involves depletion of aggrecan and breakdown of collagen fibers100. Inflammatory immune cells in the joints release proinflammatory cytokines such as IL-1β, TNF-α and IL-17. These cytokines induce expression of MMPs (MMP-1, -2, -3, -7, -8, -9 and -13) and ADAMTS (ADAMTS 1, 4 and 5) in the synovial fibroblasts and chondrocytes106107108109. Proinflammatory cytokines are the main inducers of MMPs in these cells101. In addition, there is endogenous regulation of these enzymes in synovial fibroblasts. Members of both the MMP and ADAMTS families contribute to aggrecan degradation. Collagen type II resists degradation by most proteases owing to its triple-helical structure. Only MMP-1, -8, and -13 can degrade fibrillar collagens including types I, II and III collagen110. Following cleavage of the collagen molecules, the helices are disrupted and collagen is denatured into gelatin. Gelatin then forms the substrate for gelatinases, namely MMP-2 and -99697110. Type IX and XI collagens are degraded by MMP-3 and MMP-2102. Other proteinases (cathepsins and ADAMTS) also play a role in the tissue damage in the joints96979899101111. Inhibition of these enzymes is expected to offer protection against cartilage destruction in RA. Several therapeutic interventions to treat arthritis in animal models have shown to inhibit the activities of MMPs, particularly MMP-9. For example, the treatment of arthritic rats with IL-27 or celastrol has a significant inhibitory effect on MMP-9 activity2728. (Celastrol is a bioactive component of a Chinese herb, Celastrus and other related plants). In addition, selective MMP inhibitors have been tested for their cartilage-protective effects in experimental models of arthritis112. However, there is not much information available on the inhibition of ADAMTS in RA. The development of effective and safe inhibitors of MMPs and ADAMTS would offer new therapeutic agents to limit the joint destruction.
Tissue inhibitors of metalloproteinases (TIMPs) were originally identified as endogenous proteins that regulate MMPs. However, TIMPs also can regulate ADAMTS113. Four members of the TIMPs family, namely TIMP1, 2, 3 and 4 have been identified. The expression of TIMPs is tissue specific113114. At present, there is not much information on the role of TIMPs in RA. TIMPs are also known to influence cell growth and differentiation, cell migration, and angiogenesis, and these effects are independent of their inhibitory effect on MMPs115. Imbalance in the ratio of MMPs to TIMPs promotes abnormal degradation of ECM116. Therapeutic interventions to treat arthritis in animal models have been shown to maintain the balance between MMPs and TIMPs117. For example, Sinomenine, an alkaloid derived from the Chinese medicinal plant, Sinomenium acutum, has been shown to ameliorate CIA in rats by maintaining the balance between MMPs and TIMPs117. However, the protective role of TIMPs in RA has yet to be further explored.
Influence of vascular permeability and vascular endothelial cell characteristics on target organ-directed autoimmunity
Recent studies have highlighted the role of vascular permeability of the blood vessels in the joints4 and that of the fine characteristics of the vascular endothelial cells of the joint vasculature26 in preferentially directing the systemic autoimmune responses to the joints. The K/BXN model of arthritis represents an autoimmune response against a systemic antigen, GPI. One of the mechanisms proposed for the preferential targeting of the distal joints of the paws invoked a vascular leak4. In our study on the AA model in which ubiquitously distributed Bhsp65 has been implicated in arthritis pathogenesis, we identified phage-encoded peptides that preferentially homed to the arthritic joints and showed binding to CD31+ vascular endothelial cells in the inflamed joints26. One of the two peptides also possessed anti-arthritic activity in addition to joint-homing attributes. Similarly, another study conducted in immunodeficient mice engrafted with human synovial tissue also reported isolation of synovial-binding peptides selected after the phage screening118. Taken together, these studies have opened up new avenues for further exploration of the characteristics of the joint vasculature in rendering the joints preferential targets of autoimmune attack. Additional studies are needed to determine the functional significance of unique attributes of the blood vessels as well as vascular endothelial cells in arthritis besides the much appreciated role of angiogenesis in this disease.
Therapeutic approaches for the control of autoimmune arthritis
RA is an autoimmune disease driven by proinflammatory cytokines. Therefore, if the proinflammatory cytokines can be reduced, the inflammatory component of the disease can be suppressed and the destruction of bone and cartilage limited. Several disease-modifying antirheumatic drugs (DMARDs) are now available for the management of RA patients. These medications target inflammatory mediators and are less toxic compared to other drugs for RA. One of the newer groups of anti-arthritic drugs is biologics, which are based on inhibiting the actions of proinflammatory cytokines. The first group of these drugs targets TNFα- infliximab (a chimeric monoclonal antibody), etanercept (TNFR-Fc) and adalimumab (a human monoclonal antibody)119120. The anti-TNFα therapy has worked well, but approximately 40 per cent of RA patients fail to respond to this treatment120121. Accordingly, there are drugs targeting IL-1- Anakinra (recombinant IL-1 receptor antagonist) and AMG 108 (human monoclonal antibody against IL-1β), or IL-6 receptor - Tocilizumab (a human monoclonal antibody against IL-6 receptor). There was excitement with the targeting of IL-1 because IL-1 was shown to be solely responsible for cartilage damage but partially responsible for bone damage in mice transgenic for TNFα but deficient in IL-1122. The IL-1-directed drugs have shown some efficacy in limiting inflammation, but IL-1 inhibition with Anakinra proved to have less bone damage protection than that observed with anti-TNFα treatment123.
As research on newer cytokines expands, there are increasing opportunities for new drugs to treat RA. IL-17 plays a critical role in the induction of arthritis124. Research on IL-17 has led to the development of a humanized anti-IL17 monoclonal antibody LY2439821, which has gone through phase-I clinical trials to determine patient tolerability and efficacy125. This anti-IL-17 antibody added to other DMARDs improved signs and symptoms of RA patients125. Since many cytokines signal through the Janus Kinase- Signal Transducer and Activator of Transcription (JAK-STAT) pathway, some new drugs targeting this pathway have been explored as therapeutics. For example, Tofacitinib (a JAK1 and JAK3 inhibitor), showed inhibition of the key cytokines relevant for arthritis, such as IL-17, IL-6 and IL-8126.
Angiogenesis plays an important role in the disease pathogenesis in arthritis. Several endogenous factors that have angiostatic activity are produced by the synovium of arthritic joints93 , but these factors may fail to effectively control angiogenesis and inflammation associated with arthritis. In this regard, several agents/approaches including some endogenous metabolites are being examined to control angiogenesis for eventual therapeutic use2486878889 (Table II).
Finally, complementary and alternative medicine (CAM) products are increasingly being used by arthritis patients, who are seeking alternatives to expensive and toxic conventionally used drugs. Several studies have highlighted the anti-arthritic activity of various natural plant products in experimental models of arthritis127128. For example, in studies in the AA model, extracts of green tea129, celastrus2830130, and huo-luo-xiao-ling dan131132 have been shown to offer protection against arthritis. We hope that a systematic validation of herbal products in RA patients might offer promising adjuncts to conventional drugs for the management of this debilitating disease.
Acknowledgment
This work was supported by NIH grant R01AT004321. Authors thank Eugene Kim, Rajesh Rajaiah, Yinghua Yang, Hua Yu and Siddaraju Nanjundaiah for helpful discussions.
References
- Selection of the T-cell repertoire: receptor-controlled checkpoints in T-cell development. Adv Immunol. 2004;84:201-38.
- [Google Scholar]
- The self-directed T cell repertoire: its creation and activation. Rev Immunogenet. 2000;2:26-37.
- [Google Scholar]
- Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat Immunol. 2006;7:284-92.
- [Google Scholar]
- Diversification of T cell responses to carboxy-terminal determinants within the 65-kD heat-shock protein is involved in regulation of autoimmune arthritis. J Exp Med. 1997;185:1307-16.
- [Google Scholar]
- Development of arthritis, periarthritis and periostitis in rats given adjuvants. Proc Soc Exp Biol (NY). 1956;91:95-101.
- [Google Scholar]
- Autoimmunity to chaperonins in the pathogenesis of arthritis and diabetes. Annu Rev Immunol. 1991;9:567-89.
- [Google Scholar]
- Cloning of the mycobacterial epitope recognized by T lymphocytes in adjuvant arthritis. Nature. 1988;331:171-3.
- [Google Scholar]
- Activation of T cells recognizing self 60-kD heat shock protein can protect against experimental arthritis. J Exp Med. 1995;181:943-52.
- [Google Scholar]
- Diversification of response to hsp65 during the course of autoimmune arthritis is regulatory rather than pathogenic. Immunol Rev. 1998;164:175-84.
- [Google Scholar]
- The T cells specific for the carboxyl-terminal determinants of self (rat) heat-shock protein 65 escape tolerance induction and are involved in regulation of autoimmune arthritis. J Immunol. 2004;172:2795-802.
- [Google Scholar]
- Nicotine-induced differential modulation of autoimmune arthritis in the Lewis rat involves changes in interleukin-17 and anti-cyclic citrullinated peptide antibodies. Arthritis Rheum. 2011;63:981-91.
- [Google Scholar]
- Resistance to adjuvant arthritis is due to protective antibodies against heat shock protein surface epitopes and the induction of IL-10 secretion. J Immunol. 2002;168:6463-9.
- [Google Scholar]
- Experimental autoimmune encephalomyelitis. Qualitative and semiquantitative differences in heat shock protein 60 expression in the central nervous system. J Immunol. 1995;154:3548-56.
- [Google Scholar]
- Heat shock protein 70 associations with myelin basic protein and proteolipid protein in multiple sclerosis brains. Int Immunol. 2003;15:241-9.
- [Google Scholar]
- Heat-shock proteins can promote as well as regulate autoimmunity. Autoimmun Rev. 2009;8:388-93.
- [Google Scholar]
- The K/BxN mouse model of inflammatory arthritis: theory and practice. Methods Mol Med. 2007;136:269-82.
- [Google Scholar]
- T cells against the pathogenic and protective epitopes of heat-shock protein 65 are crossreactive and display functional similarity: Novel aspect of regulation of autoimmune arthritis. J Rheumatol. 2007;34:2134-43.
- [Google Scholar]
- The regulatory C-terminal determinants within mycobacterial heat shock protein 65 are cryptic and cross-reactive with the dominant self homologs: implications for the pathogenesis of autoimmune arthritis. J Immunol. 2004;173:181-8.
- [Google Scholar]
- The determinants of susceptibility/resistance to adjuvant arthritis in rats. Arthritis Res Ther. 2009;11:239.
- [Google Scholar]
- Regulation of autoimmune arthritis by the pro-inflammatory cytokine interferon-gamma. Clin Immunol. 2008;127:98-106.
- [Google Scholar]
- Exogenous tumour necrosis factor alpha induces suppression of autoimmune arthritis. Arthritis Res Ther. 2008;10:R38.
- [Google Scholar]
- Angiogenesis inhibition as a therapeutic approach for inflammatory synovitis. Nat Clin Pract. 2007;3:434-42.
- [Google Scholar]
- Angiogenesis and vasculogenesis in rheumatoid arthritis. Curr Opin Rheumatol. 2010;22:299-306.
- [Google Scholar]
- Peptides targeting inflamed synovial vasculature attenuate autoimmune arthritis. Proc Natl Acad Sci USA. 2011;108:12857-62.
- [Google Scholar]
- Interleukin-27 and interferon-gamma are involved in regulation of autoimmune arthritis. J Biol Chem. 2011;286:2817-25.
- [Google Scholar]
- Celastrus-derived celastrol suppresses autoimmune arthritis by modulating antigen-induced cellular and humoral effector responses. J Biol Chem. 2011;286:15138-46.
- [Google Scholar]
- A cytokine-centric view of the pathogenesis and treatment of autoimmune arthritis. J Interferon Cytokine Res. 2011;31:927-40.
- [Google Scholar]
- Celastrus and its bioactive celastrol protect against bone damage in autoimmune arthritis by modulating osteoimmune cross-talk. J Biol Chem. 2012;287:22216-26.
- [Google Scholar]
- Interleukin 12 acts directly on CD4+ T cells to enhance priming for interferon gamma production and diminishes interleukin 4 inhibition of such priming. Proc Natl Acad Sci USA. 1993;90:10188-92.
- [Google Scholar]
- Differential regulation of T helper phenotype development by interleukins 4 and 10 in an alpha beta T-cell-receptor transgenic system. Proc Natl Acad Sci USA. 1992;89:6065-9.
- [Google Scholar]
- Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744-8.
- [Google Scholar]
- Breaking old paradigms: Th17 cells in autoimmune arthritis. Clin Immunol. 2009;132:295-304.
- [Google Scholar]
- Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235-8.
- [Google Scholar]
- IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484-7.
- [Google Scholar]
- Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875-86.
- [Google Scholar]
- CD8+ T cell-mediated suppression of autoimmunity in a murine lupus model of peptide-induced immune tolerance depends on Foxp3 expression. J Immunol. 2007;178:7649-57.
- [Google Scholar]
- Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537-45.
- [Google Scholar]
- Pathogenesis of rheumatoid arthritis: targeting cytokines. Ann NY Acad Sci. 2005;1051:716-29.
- [Google Scholar]
- Collagen-induced arthritis as an animal model for rheumatoid arthritis: focus on interferon-gamma. J Interferon Cytokine Res. 2011;31:917-26.
- [Google Scholar]
- Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 1989;2:244-7.
- [Google Scholar]
- Stimulation of bone resorption and inhibition of bone formation in vitro by human tumour necrosis factors. Nature. 1986;319:516-8.
- [Google Scholar]
- IL-1 alpha beta blockade prevents cartilage and bone destruction in murine type II collagen-induced arthritis, whereas TNF-alpha blockade only ameliorates joint inflammation. J Immunol. 1999;163:5049-55.
- [Google Scholar]
- Interleukin-1beta and interleukin-6 in arthritis animal models: roles in the early phase of transition from acute to chronic inflammation and relevance for human rheumatoid arthritis. Mol Med. 2010;16:552-7.
- [Google Scholar]
- Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. 1998;161:409-14.
- [Google Scholar]
- Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146-55.
- [Google Scholar]
- Interleukin-17 is produced by both Th1 and Th2 lymphocytes, and modulates interferon-gamma- and interleukin-4-induced activation of human keratinocytes. J Invest Dermatol. 2000;115:81-7.
- [Google Scholar]
- RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol. 2011;12:320-6.
- [Google Scholar]
- Interleukin 17 levels are increased in juvenile idiopathic arthritis synovial fluid and induce synovial fibroblasts to produce proinflammatory cytokines and matrix metalloproteinases. J Rheumatol. 2008;35:515-9.
- [Google Scholar]
- Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673-82.
- [Google Scholar]
- Interleukin-27 priming of T cells controls IL-17 production in trans via induction of the ligand PD-L1. Immunity. 2012;36:1017-30.
- [Google Scholar]
- Up-regulation of stromal cell-derived factor 1 (CXCL12) production in rheumatoid synovial fibroblasts through interactions with T lymphocytes: role of interleukin-17 and CD40L-CD40 interaction. Arthritis Rheum. 2007;56:1076-86.
- [Google Scholar]
- Interleukin-17 regulates expression of the CXC chemokine LIX/CXCL5 in osteoblasts: implications for inflammation and neutrophil recruitment. J Leukoc Biol. 2004;76:135-44.
- [Google Scholar]
- IL-17 contributes to angiogenesis in rheumatoid arthritis. J Immunol. 2010;184:3233-41.
- [Google Scholar]
- Exacerbation of antigen-induced arthritis in IFN-gamma-deficient mice as a result of unrestricted IL-17 response. J Immunol. 2007;179:6228-36.
- [Google Scholar]
- WSX-1 and glycoprotein 130 constitute a signal-transducing receptor for IL-27. J Immunol. 2004;172:2225-31.
- [Google Scholar]
- A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol. 2007;8:1380-9.
- [Google Scholar]
- Interleukin-27 inhibits human osteoclastogenesis by abrogating RANKL-mediated induction of nuclear factor of activated T cells c1 and suppressing proximal RANK signaling. Arthritis Rheum. 2010;62:402-13.
- [Google Scholar]
- Immunisation against heterologous type II collagen induces arthritis in mice. Nature. 1980;283:666-8.
- [Google Scholar]
- Temporal expression of inflammatory cytokines and chemokines in rat adjuvant-induced arthritis. Arthritis Rheum. 2000;43:1266-77.
- [Google Scholar]
- Cytokine expression and synovial pathology in the initiation and spontaneous resolution phases of adjuvant arthritis: interleukin-17 expression is upregulated in early disease. Clin Exp Immunol. 2001;123:487-95.
- [Google Scholar]
- The dynamics of articular leukocyte trafficking and the immune response to self heat-shock protein 65 influence arthritis susceptibility. J Clin Immunol. 2008;28:420-31.
- [Google Scholar]
- Antibody responses to mycobacterial and self heat shock protein 65 in autoimmune arthritis: epitope specificity and implication in pathogenesis. J Immunol. 2006;177:6634-41.
- [Google Scholar]
- Regulation of autoimmune inflammation by pro-inflammatory cytokines. Immunol Lett. 2008;120:1-5.
- [Google Scholar]
- Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999;162:2347-52.
- [Google Scholar]
- IL-17 induces hyperalgesia via TNF-dependent neutrophil infiltration. Pain. 2011;152:1838-45.
- [Google Scholar]
- Synovial biology and T cells in rheumatoid arthritis. Pathophysiology. 2005;12:183-9.
- [Google Scholar]
- Chemokines and chemokine receptors in rheumatoid arthritis. Semin Immunol. 2003;15:15-21.
- [Google Scholar]
- Suppression of autoimmune arthritis by Celastrus-derived Celastrol through modulation of pro-inflammatory chemokines. Bioorg Med Chem. 2012;20:5229-34.
- [Google Scholar]
- Angiogenesis in rheumatoid arthritis: a disease specific process or a common response to chronic inflammation? Autoimmun Rev. 2011;10:595-8.
- [Google Scholar]
- The vasculature in rheumatoid arthritis: cause or consequence? Int J Exp Pathol. 2009;90:249-61.
- [Google Scholar]
- Vascular endothelial growth factor expression and regulation of murine collagen-induced arthritis. J Immunol. 2000;164:5922-7.
- [Google Scholar]
- Norisoboldine, an alkaloid compound isolated from Radix linderae, inhibits synovial angiogenesis in adjuvant-induced arthritis rats by moderating Notch1 pathway-related endothelial tip cell phenotype. Exp Biol Med (Maywood). 2012;237:919-32.
- [Google Scholar]
- Suppression of collagen-induced arthritis by an angiogenesis inhibitor, AGM-1470, in combination with cyclosporin: reduction of vascular endothelial growth factor (VEGF) Cell Immunol. 1995;166:196-206.
- [Google Scholar]
- Treatment of collagen-induced arthritis with recombinant plasminogen-related protein B: a novel inhibitor of angiogenesis. J Orthop Sci. 2011;16:443-50.
- [Google Scholar]
- Arginine-rich anti-vascular endothelial growth factor (anti-VEGF) hexapeptide inhibits collagen-induced arthritis and VEGF-stimulated productions of TNF-alpha and IL-6 by human monocytes. J Immunol. 2005;174:5846-55.
- [Google Scholar]
- Regulation of cytokine-induced HIF-1alpha expression in rheumatoid synovial fibroblasts. Ann NY Acad Sci. 2007;1108:340-8.
- [Google Scholar]
- Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum. 2002;46:2587-97.
- [Google Scholar]
- An angiogenesis inhibitor, 2-methoxyestradiol, involutes rat collagen-induced arthritis and suppresses gene expression of synovial vascular endothelial growth factor and basic fibroblast growth factor. J Rheumatol. 2008;35:2119-28.
- [Google Scholar]
- New antiangiogenic strategies for the treatment of proliferative synovitis. Expert Opin Investig Drugs. 2005;14:1-17.
- [Google Scholar]
- Treatment with soluble VEGF receptor reduces disease severity in murine collagen-induced arthritis. Lab Invest. 2000;80:1195-205.
- [Google Scholar]
- Angiogenesis inhibitors for the treatment of chronic autoimmune inflammatory arthritis. Curr Opin Investig Drugs. 2009;10:425-33.
- [Google Scholar]
- Mechanisms of adaptive angiogenesis to tissue hypoxia. Angiogenesis. 2008;11:121-40.
- [Google Scholar]
- Angiogenesis and its targeting in rheumatoid arthritis. Vascul Pharmacol. 2009;51:1-7.
- [Google Scholar]
- Hypoxia augments cytokine (transforming growth factor-beta (TGF-beta) and IL-1)-induced vascular endothelial growth factor secretion by human synovial fibroblasts. Clin Exp Immunol. 1999;115:176-82.
- [Google Scholar]
- Expression of vascular endothelial growth factor in synovial fibroblasts is induced by hypoxia and interleukin 1beta. J Rheumatol. 1997;24:1253-9.
- [Google Scholar]
- Collagenase, cathepsin B and cathepsin L gene expression in the synovial membrane of patients with early inflammatory arthritis. Rheumatology (Oxford). 1999;38:34-42.
- [Google Scholar]
- Collagenase activity of cathepsin K depends on complex formation with chondroitin sulfate. J Biol Chem. 2002;277:28669-76.
- [Google Scholar]
- Degradation of cartilage matrix components by the cysteine proteinases, cathepsins B and L. Biomed Biochim Acta. 1991;50:561-4.
- [Google Scholar]
- Matrix metalloproteinases (MMPs) in health and disease: an overview. Front Biosci. 2006;11:1696-701.
- [Google Scholar]
- Proteinases in the joint: clinical relevance of proteinases in joint destruction. Arthritis Res Ther. 2007;9:221.
- [Google Scholar]
- Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827-39.
- [Google Scholar]
- Fractalkine: a novel angiogenic chemokine in rheumatoid arthritis. Am J Pathol. 2001;159:1521-30.
- [Google Scholar]
- Role of the CCL21 and CCR7 pathways in rheumatoid arthritis angiogenesis. Arthritis Rheum. 2012;64:2471-81.
- [Google Scholar]
- Type II collagen degradation and its regulation in articular cartilage in osteoarthritis. Ann Rheum Dis. 2002;61(Suppl 2):ii78-81.
- [Google Scholar]
- Stimulation of the secretion of latent cysteine proteinase activity by tumor necrosis factor alpha and interleukin-1. Arthritis Rheum. 1993;36:772-80.
- [Google Scholar]
- Expression of proteinases and inflammatory cytokines in subchondral bone regions in the destructive joint of rheumatoid arthritis. Rheumatology (Oxford). 2001;40:247-55.
- [Google Scholar]
- The role of interleukin-17 in mediating joint destruction in rheumatoid arthritis. Biochem Biophys Res Commun. 2010;397:131-5.
- [Google Scholar]
- Contemporary concepts of inflammation, damage and repair in rheumatic diseases. Best Pract Res Clin Rheumatol. 2006;20:829-48.
- [Google Scholar]
- Expression of MMPs and TIMPs genes in human breast cancer epithelial cells depends on cell culture conditions and is associated with their invasive potential. Anticancer Res. 2004;24:4025-30.
- [Google Scholar]
- Molecular cloning of a possible cysteine proteinase predominantly expressed in osteoclasts. J Biol Chem. 1994;269:1106-9.
- [Google Scholar]
- Joint diseases and matrix metalloproteinases: a role for MMP-13. Curr Pharm Biotechnol. 2008;9:47-54.
- [Google Scholar]
- Molecular cloning and characterization of human tissue inhibitor of metalloproteinase 4. J Biol Chem. 1996;271:30375-80.
- [Google Scholar]
- The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 2010;1803:55-71.
- [Google Scholar]
- Imbalance between interstitial collagenase and tissue inhibitor of metalloproteinases 1 in synoviocytes and fibroblasts upon direct contact with stimulated T lymphocytes: involvement of membrane-associated cytokines. Arthritis Rheum. 1998;41:1748-59.
- [Google Scholar]
- Sinomenine ameliorates arthritis via MMPs, TIMPs, and cytokines in rats. Biochem Biophys Res Commun. 2008;376:352-7.
- [Google Scholar]
- Identification of synovium-specific homing peptides by in vivo phage display selection. Arthritis Rheum. 2002;46:2109-20.
- [Google Scholar]
- Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheum. 1993;36:1681-90.
- [Google Scholar]
- Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum. 1998;41:1552-63.
- [Google Scholar]
- TNF-induced structural joint damage is mediated by IL-1. Proc Natl Acad Sci USA. 2007;104:11742-7.
- [Google Scholar]
- Updated consensus statement on biological agents, specifically tumour necrosis factor {alpha} (TNF{alpha}) blocking agents and interleukin-1 receptor antagonist (IL-1ra), for the treatment of rheumatic diseases, 2005. Ann Rheum Dis. 2005;64(Suppl 4):iv2-14.
- [Google Scholar]
- IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acad Sci USA. 2003;100:5986-90.
- [Google Scholar]
- LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum. 2010;62:929-39.
- [Google Scholar]
- In vitro and in vivo analysis of a JAK inhibitor in rheumatoid arthritis. Ann Rheum Dis. 2012;71(Suppl 2):i70-4.
- [Google Scholar]
- Herbal medicinal products target defined biochemical and molecular mediators of inflammatory autoimmune arthritis. Bioorg Med Chem. 2011;19:21-9.
- [Google Scholar]
- Immunomodulation of autoimmune arthritis by herbal CAM. Evid Based Complement Alternat Med 2011 2011 986797
- [Google Scholar]
- Green tea protects rats against autoimmune arthritis by modulating disease-related immune events. J Nutr. 2008;138:2111-6.
- [Google Scholar]
- Celastrus aculeatus Merr. suppresses the induction and progression of autoimmune arthritis by modulating immune response to heat-shock protein 65. Arthritis Res Ther. 2007;9:R70.
- [Google Scholar]
- Suppression of ongoing experimental arthritis by a chinese herbal formula (huo-luo-xiao-ling dan) involves changes in antigen-induced immunological and biochemical mediators of inflammation. Evid Based Complement Alternat Med 2011 2011 642027
- [Google Scholar]
- Modified huo-luo-xiao-ling dan suppresses adjuvant arthritis by inhibiting chemokines and matrix-degrading enzymes. Evid Based Complement Alternat Med 2012 2012 589256
- [Google Scholar]




