
Translate this page into:
Safety analysis of all active risk evaluation & mitigation strategy for U.S. FDA approved drugs – An Indian perspective
*For correspondence: renuka.munshi@gmail.com
-
Received: ,
Sir,
Medication risk management is an important factor considered by all national drug regulatory authorities when approving drugs. Some initiatives were taken by the drug regulatory authorities in the past such as the Durham Humphrey Amendment (1951), which assigned product designation as a prescription drug and refrained from labelling it as over-the-counter (OTC) drug to ensure that certain drugs are administered only under supervision of a healthcare physician and distributed with appropriate medical prescription1,2. Before 2007, the United States Food and Drug administration (U.S. FDA) would occasionally ask companies to have a special drug safety plan called Risk Management Programmes (RMPs) to mitigate serious risks associated with those drug products that provided substantial therapeutic benefits. Following United States Food and Drug Administration Amendments Act (FDAAA) 2007, the U.S. FDA received the authority to ask for risk mitigation/minimisation strategy from the manufacturers to ensure that the benefits of a drug/biological product outweigh the associated safety concerns. A risk evaluation and mitigation strategy (REMS) is one such U.S. FDA initiative to maximize the benefit of drugs consumed by individuals with precautions to be followed to minimize the adverse effects3. REMS are designed not only for reinforcing medication use behaviours but also to instil actions among the end users that can support the safe use of those medicines and in turn reduce the occurrence and/or severity of certain serious risks, as described in the FDA-approved prescription information for the given medications. Almost all medications come with labelling information about medication risks for healthcare stakeholders, however, a few medications do require a REMS in place. The four types of key elements of REMS issued by the U.S. FDA are: (i) ‘Elements To Assure Safe Use’ (ETASU) that require certified clinicians or healthcare providers to prescribe and participate in training, patient counselling and monitoring; (ii) communication plan (CP) may include informational material with letters, websites and fact sheets providing specific safety risks listed in REMS; (iii) medication guide (MG) is an element that may be provided separately with the medicines or can be part of their labelling; and (iv) REMS may be issued both as a ‘communication plan’ as well as a medication guide4. Every new REMS proposal for new drug application (NDA) or biologics license application (BLA) should typically also include the REMS assessment reports for U.S. FDA review. REMS focuses on monitoring and/or managing and preventing a specific adverse risk associated with a particular drug product, by means of educating and/or reinforcing actions so as to reduce the frequency and/or severity of the adverse events thereby facilitating the intent of pharmacovigilance programme globally for safety reporting3.
In this study a safety analysis of all U.S. FDA-approved REMS was performed to evaluate the nature of drug-related toxicity and number of REMS issued by U.S. FDA year-wise as well as in each therapeutic domain. The U.S. FDA website was accessed for data (https://www.accessdata.fda.gov/scripts/cder/rems/index.cfm) and the last updated active REMS data up to September 6, 2022 were extracted for analysis5. The search strategy involved individually screening each drug detail for its goals to know the safety concern associated with each drug at REMS U.S. FDA database. Some drugs had more than one type of toxicity risk for which REMS was issued and this was categorized separately under the respective therapeutic domain as per the organ system affected. If more than one safety concern was related to the same organ system for a given drug, then it was calculated only once. The data collected included the drug name approved for REMS with their approval dates, drug-related toxicity for which REMS was approved by U.S. FDA, elements of REMS and year-wise distribution of REMS that was issued. Descriptive statistics was used for analyses and the results were summarized in proportions/percentages.
Our study analyses revealed that there were 60 active REMS issued by the U.S. FDA for drugs used in various therapeutic areas as of September 6, 2022. Among the total 60 active REMS issued by the U.S. FDA that were analyzed, 55 (92%) included ETASU, 3 (5%) included only a CP REMS element, 1 (2%) included only the ‘medication guide’ REMS element while 1 (2%) included the CP as well as the ‘medication guide’ REMS elements. These 60 active REMS issued by the U.S. FDA were evaluated for the nature of drug-related toxicity and the number of REMS issued in each therapeutic domain. However, a single drug may have had one REMS issued for more than one safety concern, ranging from a minimum of one to a maximum of four safety concerns; hence the absolute number of drug-related issues was assessed descriptively (Fig. 1).
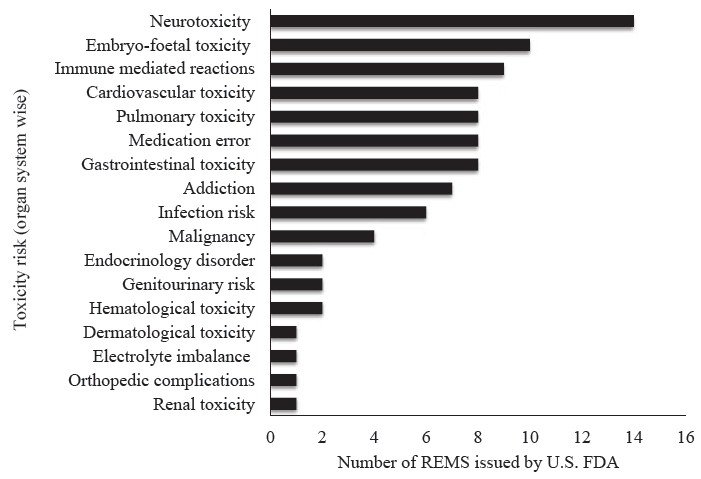
- Nature of drug-related toxicity and number of REMS issued in respective domains. REMS, Risk Evaluation and Mitigation Strategies; FDA, Food & Drug Administration.
Observations from this study revealed that the maximum REMS were issued for drug-related neurotoxicity (n=14) followed by embryo-foetal toxicity (n=10) and immune-mediated toxicity (n=9). Immune-mediated reactions mainly included risk for cytokine release syndrome, autoimmune reaction, infusion reaction and anaphylaxis. There was one REMS issued for psychiatric suicidal ideation risk with brodalumab injection, for cutaneous reaction with the drug duvelisib, sudden death risk with vandetanib due to QT interval prolongation, glomerulonephritis with injection inotersen, osteonecrosis of jaw/atypical femoral fracture with intravenous denosumab and one medical device–related alert with the use of buprenorphine hydrochloride implant prone to migration, protrusion and expulsion. Buprenorphine had REMS issued for three formulations including trans-mucosal route, extended-release injection and as an implant device. Similarly, more than one REMS were issued for alosetron-2, sodium oxybate-2 and opioid analgesics-3. Medication errors included those with the risk of drug administration errors and accidental drug overdose toxicity. Of these, 60 active REMS-approved drugs, only 19 (31.67%) were approved by the Central Drugs Standard Control Organization (CDSCO), drug regulatory authority of India. When year-wise active REMS data (n=60) were analyzed, maximum REMS (10%) were issued in years 2010, 2012, 2017 and 2018 (Fig. 2).

- Year-wise distribution of REMS approved by U.S. FDA.
U.S. FDA launched a list of medications with an approved REMS on public domain database on December 17, 2021. All drugs approved with benefits always come at the cost of some side effects. Hence, risk evaluation and mitigation strategies become a necessary component of drug administration and minimizing those risks depend on different stakeholders linked to drugs-associated REMS which may include patients, healthcare professionals and pharmacists who dispense or administer these drugs. Creating awareness among these stakeholders forms an important component for the implementation of REMS and the drug approval process. The REMS guidance document for industry provides updated recommendations on the format as well as the content of REMS for a particular prescription drug product, including a biological drug product6. REMS Industry guidance (2019) was revised to provide information on how the U.S. FDA defines, processes and accepts the modifications made to already approved REMS. Specifically, this guidance provides information, mentioned in the Section 505-1(h) of the Federal Food, Drug, and Cosmetic FD&C Act, which is on the type of changes to REMS that will be considered as modifications or revisions of REMS7. The draft guidance of REMS Assessment: Planning and Reporting (February 2019) recommends assessing the REMS using both process as well as outcome measure metrics to evaluate the performance of REMS8. Furthermore, the United States, Secretary of Health and Human Services (HHS) issued the guidance (2020) to communicate its temporary policy for certain REMS requirements for the duration of any public health emergency (PHE)9. In our study, neurotoxicity risk followed by embryo foetal toxicity was most common for which REMS was issued similar to the embryo foetal toxicity reported in the study findings by Boudes et al10 (2017). In India, there is no concept of REMS and information on medication risks is simply labelled with warnings. However, from an Indian standpoint too, the CDSCO has now begun insisting for REMS during joint inspection of pharmaceutical plants during WHO Good Manufacturing audits. While our regulators have initiated the need to submit the REMS information, U.S. FDA can be seen revising REMS to develop new steps to ensure safety strategy. The Indian drug regulators have issued a pharmacovigilance guidance document (effective from January 2018) for marketing authorization holders of pharmaceutical products. Though there exists a lag in the drug approval process variable across the countries, REMS can be issued for the approved drugs as well as for those under process in India with maximum drug consumers across the world. Few limitations still exist with REMS. It cannot prevent all risks but will help to mitigate it and needs to be updated periodically with evolving new drug information.
The risk management process for drug safety is equally important. It should involve a risk assessment plan (risk identification, analysis and evaluation), risk control, risk communication and risk review. REMS is an important measure to mitigate medication risk and ensure public health safety. However, there is a need to develop an Indian harmonized guidance document for its implementation in a more effective manner across the world.
Financial support & sponsorship
None.
Conflicts of Interest
None.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing of the manuscript and no images were manipulated using AI.
References
- History of Federal Regulation: 1902–Present. Available from: https://www.fdareview.org/issues/history-of-federal-regulation-1902-present/#p06, accessed on September 16, 2022
- Roadmap to risk evaluation and mitigation strategies (REMS) success. Ther Adv Drug Saf. 2010;1:21-38.
- [CrossRef] [PubMed] [Google Scholar]
- Risk Evaluation and Mitigation Strategies/REMS. Available from: https://www.fda.gov/drugs/drug-safety-and-availability/risk-evaluation-and-mitigation-strategies-rems, accessed on September 16, 2022.
- Approved Risk Evaluation and Mitigation Strategies (REMS). REMS reports. Available from: https://www.accessdata.fda.gov/scripts/cder/rems/index.cfm?event=RemsData.page, accessed on September 16, 2022.
- Approved Risk Evaluation and Mitigation Strategies (REMS). REMS @FDA. Available from: https://www.accessdata.fda.gov/scripts/cder/rems/index.cfm, accessed on September 16, 2022.
- Format and content of a REMS document guidance for industry. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/format-and-content-rems-document-guidance-industry, accessed on February 16, 2024.
- Risk Evaluation and Mitigation Strategies Modifications and Revisions Guidance for Industry. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/risk-evaluation-and-mitigation-strategies-modifications-and-revisions-guidance-industry, accessed on September 16, 2022.
- REMS Assessment: Planning and Reporting, Draft guidance for Industry. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/rems-assessment-planning-and-reporting, accessed on September 16, 2022.
- Policy for Certain REMS Requirements during the COVID-19 Public Health Emergency Guidance for Industry and Health Care Professionals. Available from: https://www.supremecourt.gov/opinions/URLs_Cited/OT2020/20A34/20A34-5.pdf, accessed on September 16, 2022.
- Risk Evaluation and Mitigation Strategies (REMSs): Are they improving drug safety? A Critical Review of REMSs Requiring Elements to Assure Safe Use (ETASU) Drugs RD. 2017;17:245-254.
- [CrossRef] [Google Scholar]




