Translate this page into:
Novel mutations in PRG4 gene in two Indian families with camptodactyly-arthropathy- coxa vara- pericarditis (CACP) syndrome
Reprint requests: Dr Prajnya Ranganath, Assistant Professor & Head, Department of Medical Genetics Nizam's Institute of Medical Sciences, Hyderabad 500 082, India e-mail: prajnyaranganath@gmail.com
-
Received: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
Camptodactyly – arthropathy- coxa vara- pericarditis (CACP) syndrome is an autosomal recessive disorder caused by mutations in the PRG4 (proteoglycan 4) gene. Hallmarks of the syndrome include congenital or early-onset camptodactyly and arthropathy with synovial hyperplasia, progressive coxa vara deformity and non-inflammatory pericardial effusions. Till date only around 25 pathogenic mutations have been reported in this gene and none have been reported from India. We report here the mutations in the PRG4 gene in three patients of CACP from two unrelated families from India.
Methods:
Molecular genetic studies were done for the three patients with the CACP syndrome, from two unrelated Indian families, through sequence analysis of all coding exons and the exon-intron boundaries of the PRG4 gene.
Results:
Two novel frame-shift deletion mutations leading to premature protein termination were found. One patient was identified to be homozygous for a 2 base pair deletion in exon 6 (c.2645_2646delGA) and the two affected siblings from the other family were found to be homozygous for a 4 base pair deletion in exon 6 (c.2883_2886delAAGA).
Conclusions:
This is perhaps the first report of PRG4 mutations from India. Further mutation studies in Indian CACP cases will help to determine the mutation spectrum of the PRG4 gene in the Indian population and also help to further elucidate the molecular pathology and the genotype-phenotype correlation of this rare disease.
Keywords
CACP syndrome
Indian cases
novel mutations
PRG4 gene

Camptodactyly-arthropathy-coxa vara- pericarditis (CACP) syndrome (OMIM # 208250) is a rare autosomal recessive condition characterized by the association of congenital or early onset camptodactyly and non-inflammatory arthropathy with synovial hyperplasia. Progressive coxa vara deformity and/or non-inflammatory pericardial or pleural effusions are also observed in many patients with this condition12. Using homozygosity mapping, the CACP locus was assigned to a 1.9 cM interval on the human chromosome 1q25-313. Later, using the positional cloning approach, the causative gene for CACP was identified to be the proteoglycan 4 gene (PRG4; OMIM# 604283; Refseq: NM- 005807)4.
Till date only 15 disease causing mutations in the PRG4 gene have been listed in the Human Genome Mutation Database (www.hgmd.cf.ac.uk), in CACP patients of Caucasian, Egyptian, Saudi Arabian, Korean, UAE and Pakistani origin. Each of these reported disease-causing mutations has been predicted to prematurely truncate the protein product4567. In addition, there are two recent reports, one of five novel mutations and the first case of uniparental disomy in a European cohort of CACP cases and another of an additional five novel mutations in Saudi Arabian patients with CACP89. Mutational spectrum of the PRG4 gene in Indian cases of CACP has not been reported. Here we report the clinical and molecular genetic findings of three patients with CACP from two unrelated Indian families. In addition, the study also reports two novel, hitherto unreported mutations in the PRG4 gene, which further expands the known spectrum of disease-causing mutations in the gene.
Material & Methods
Three affected individuals from two unrelated families were clinically diagnosed to have the CACP syndrome and were included in the study. Of the three patients, two were siblings aged 10 and 5 yr, respectively (both male) (patients 1a and 1b) and the third was a 16 yr old girl (patient 2) from an unrelated family. The two affected brothers belonged to a Hindu family from the Katihar district of Bihar. They were the second and the fifth offspring respectively of non-consanguineous parents; their other three siblings were normal. They were clinically evaluated and diagnosed to have the CACP syndrome at the Medical Genetics department of the Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, India in January 2009. Both siblings presented with a history of persistent painless swelling involving the knee, ankle, elbow and wrist joints and contractures involving the small joints of the hands and feet since around two years of age. Neither of them had any history of rashes or recurrent fever. Both had a normal developmental history. The only salient findings on physical examination were bilateral symmetric non-tender elbow, wrist, knee and ankle joint effusions with synovial thickening and flexion contractures of bilateral wrist, interphalangeal, metacarpophalangeal and metatarsophalangeal joints in both (Fig. 1A & B). Skeletal survey revealed increased joint spaces suggestive of effusion in the knee, elbow and wrist joints with periarticular severe osteopenia especially in the elbow joints along with soft tissue swelling (Fig. 1C & D). The female patient was 16 yr old and was the first offspring of non-consanguineous parents, belonging to a Hindu family from the Nalgonda district of Andhra Pradesh. She was clinically evaluated and diagnosed to have the CACP syndrome at the Medical Genetics department of the Nizam's Institute of Medical Sciences, Hyderabad, Andhra Pradesh, India, in December 2011. She presented with a history of persistent painless swelling involving the knee, ankle and elbow joints and contractures involving the small joints of the hands since early childhood. Her developmental milestones were normal and she did not have any other systemic symptoms. There was no history of any other similarly affected individuals in her family. The salient findings on physical examination were bilateral symmetric non-tender elbow, knee and ankle joint effusions with synovial thickening and contractures of bilateral wrist, interphalangeal, metacarpophalangeal and metatarsophalangeal joints. Skeletal survey in this patient revealed increased joint spaces suggestive of effusion in the knee and elbow joints with periarticular osteopenia.
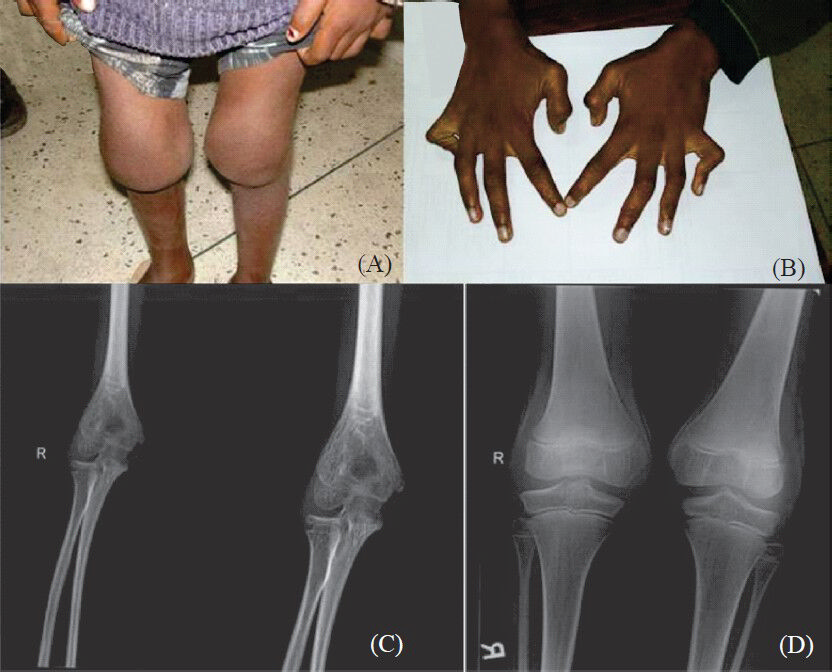
-
(A). Photograph of the knee joints of patient 1b showing bilateral knee joint swelling. (B) Hands of patient 1a showing contractures of the metacarpophalangeal and interphalangeal joints. (C) Elbow radiograph of patient 1a showing widening of the joint space and severe periarticular osteopenia. (D) Knee radiograph of patient 1a showing widening of the joint space and periarticular osteopenia.
Though all three patients were initially misdiagnosed to have juvenile rheumatoid arthritis at other centers, they were clinically suspected to have CACP syndrome on the basis of the painless joint effusions, arthropathy and camptodactyly. None of the three patients had pericarditis. However, the absence of pericarditis does not rule out this syndrome as in some cases pericarditis has been reported to be absent or to set in later in the course of the disease2.
DNA extraction & sequencing: The approval of the Institute Ethics Committee of the Centre for DNA Fingerprinting and Diagnostics was obtained for the study. As all three patients were minors, informed written consent was procured from their parents for the study. Five ml of peripheral blood (anticoagulated with potassium - EDTA) was collected from each of the three patients following the clinical diagnosis. In addition, 5 ml peripheral blood samples (anticoagulated with potassium - EDTA) were also collected from the parents of the two affected siblings (Patients 1a and 1b). Genomic DNA was extracted from the whole blood samples using the standard salting out method. PCR primers were designed to amplify the coding sequences and intron-exon boundaries of the candidate gene using Primer 3 input (version 0.4.0) tool (http://www.primer3plus.com/web_0.4.0/input.htm; accessed on February 12, 2012) (Table). Polymerase chain reaction (PCR) was performed using the Eppendorf Mastercycler gradient (Eppendorf Scientific, Hauppauge, NY; www.eppendorf.com). For amplification of PRG4 from genomic DNA, the PCR mixture (12.5 μl) contained 100 mM Tris HCl (pH 8.8), 500 mM KCl, 0.8% (v/v) Nonidet P40, 10 mm dNTPs, 25 mM MgCl2, 100 ng of genomic DNA, 10 pmol of the forward and reverse primers, and 5 units Taq polymerase (Fermentas Life Sciences, Ontario, Canada). The cycling conditions included an initial denaturation at 94°C for 4 min followed by 30 cycles at 94°C for 30 sec, 55-65°C for 55 sec and 72°C for 55 sec with a final extension at 72°C for 5 min. The PCR products were resolved by agarose gel electrophoresis. The PCR products were purified using QIAquick PCR purification kit (Qiagen, Hilden, Germany) and sequenced on ABI prism 377 automated DNA sequencer (Applied Biosystems, USA).
Mutation detection: Sequence data were analyzed using the chromasliteV 2.01 software (http://www.technelysium.com.au/chromas_lite.html) and aligned with reference Genbank cDNA sequence NM_005807.2 using Clustal W multiple sequence alignment tools (http://www.ebi.ac.uk/tools/psa/emboss_needle/nucleotide.html).The disease-causing potential of sequence alterations was evaluated using the Mutation taster software tool (http://www.mutationtaster.org). The mutations were checked with already available PRG4 gene mutations from the Human Gene Mutation Database (www.hgmd.cf.ac.uk).
Results
The entire coding region of the PRG4 gene was assessed in our patients through bidirectional DNA sequencing, with the exception of a 1702 bp region in exon 6 which was not amenable to amplification due to its highly repetitive character. Analysis revealed two novel frame-shift mutations in our CACP patients. The affected siblings (patients 1a and 1b) were found to be homozygous for a 4 base pair deletion in exon 6 (c.2883_2886delAAGA; p.Glu961glufsX39) (Fig. 2A). Molecular genetic testing of their parents confirmed that each of them was a heterozygous carrier of this 4 base pair deletion mutation. The female patient (patient 2) was found to be homozygous for a 2 base pair deletion in exon 6 (c.2645_2646delGA; p.Asp882LysfsX5) (Fig. 2B). Parental testing to confirm their heterozygous status for this deletion mutation could not be done for patient 2, as the parental samples in this case could not be procured. The disease-causing potential of sequence alterations was evaluated using the Mutation taster software tool. The 4 base pair deletion affecting Glu 961 in patients 1a and 1b predicts premature termination after 39 residues whereas the 2 base pair deletion affecting Asp882 in patient 2 predicts premature termination five residues later. Further proof for the pathogenicity of the two detected mutations was obtained using the Mutation Taster software tool, which predicted that both mutations were ‘disease-causing’.
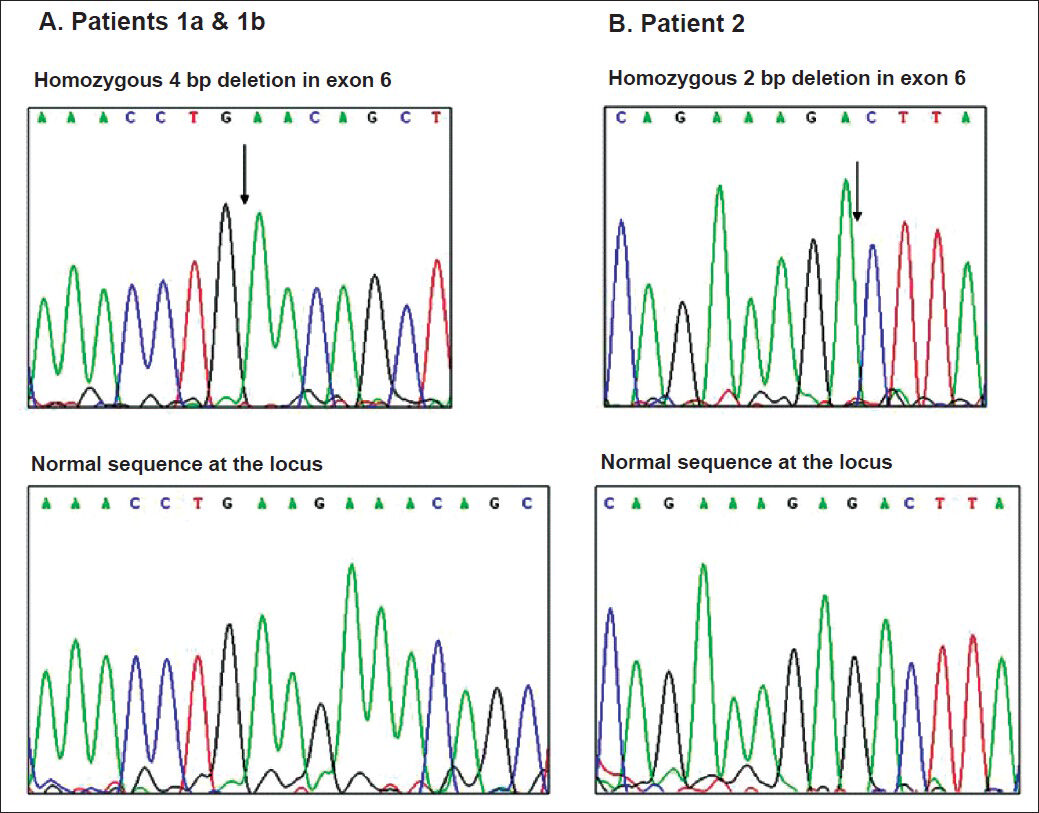
- Electropherogram of wild type alleles (top) and mutant alleles (bottom) of patients 1a and 1b and patient 2. The sites of homozygous deletion are indicated by arrows.
Discussion
The PRG4 gene is the only gene known to be associated with the camptodactyly-arthropathy-coxa vara- pericarditis syndrome. Among the products of full length PRG4 transcripts is a secreted glycoprotein of 1404 amino acids termed lubricin, a major constituent of synovial joints10. Lubricin is a large proteoglycan that functions as the major lubricant in articulating joints, produced and secreted by synoviocytes and superficial zone chondrocytes1112. It is present in the synovial fluid and at cartilage surfaces1314. Functions attributed to lubricin within articulating joints include protection of cartilage surfaces from protein deposition and cell adhesion, inhibition of synovial cell growth, and boundary lubrication at the articular cartilage surface15. Lubricin contains multiple protein domains namely somatomedin B (SMB), hemopexin-like (PEX) domain and a large, central extensively O- linked glycosylated, mucin-like domain that likely contribute to its diverse biologic properties1516. Studies involving synovial fluid from patients with the CACP syndrome have suggested that synovial fluid lacking lubricin would be less able to dissipate the energy of impact on the joints that occurs during locomotion17.
Individuals with CACP have normal appearing joints at birth, but with advancing age develop joint pathology associated with non-inflammatory synoviocyte hyperplasia and sub-intimal fibrosis of the synovial capsule3. These patients also develop pericarditis and finger joint flexion contractures, likely due to insufficient surface lubrication between the visceral and parietal pericardium and the tendon and tendosynovium respectively. Initial studies have shown that the genetic disruption of the PRG4 gene in mouse recapitulates clinical features of CACP in humans15.
Since the identification of PRG4 as the causative gene for CACP by Marcelino and co-workers in 19994, 15 mutations have been reported in the Human Genome Mutation Database (www.hgmd.cf.ac.uk) in CACP patients of Caucasian, Egyptian, Saudi Arabian, Korean, UAE and Pakistani origin567. There are two recent reports of an additional 10 novel mutations in the PRG4 gene; five each in a European cohort and a Saudi Arabian cohort of CACP cases89.
Both of our patients were found to have mutations in exon 6 of the PRG4 gene. Many of the previously reported mutations in the PRG4 gene have also been found to occur within exon 6 or an adjacent intronic splice acceptor site. This could be probably accounted for by the fact that exon 6 accounts for around 67 per cent of the coding sequence of the PRG4 gene3. It is also interesting to note that no mutations have been documented so far in exons 1-5 of this gene. This may be due to the presence of alternatively-spliced PRG4 isoforms that exclude exons 2, 4 and 5, thus masking mutations with no phenotypic consequence18. Earlier reports have also led to the conclusion that missense mutations or in frame deletions are not pathogenic and CACP syndrome occurs due to a null phenotype (i.e. premature truncation)4567. Our findings also support the previous reports that only frame shift and non-sense mutations in the PRG4 gene trigger the phenotype.
It is of interest to note that though neither of the two families gave a history of parental consanguinity, homozygosity for the respective mutations was found in both families. It is known that even in the absence of actual consanguinity, inbreeding can lead to homozygosity for recessive mutations and thereby result in disease in the offspring19. This is not an uncommon phenomenon in many regions of India where inbreeding within small communities in quite prevalent. In each of the two families, both the husband and the wife hailed from the same village and belonged to the same religious and caste group.
Acknowledgment
Authors thank the patients and their families for participating in this study, and acknowledge the Indian Council of Medical Research (ICMR), New Delhi, India, for funding and thereby enabling banking of DNA samples of Patients 1a and 1b and their parents at the Department of Medical Genetics in the Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, Uttar Pradesh, India.
References
- A syndrome of fibrosing pleuritis, pericarditis, and synovitis with infantile contractures of fingers and toes in 2 sisters: ‘familial fibrosing serositis’. J Rheumatol. 1995;22:2349-55.
- [Google Scholar]
- Clinical variability and genetic homogeneity of the camptodactyly-arthropathy-coxa vara-pericarditis syndrome. Am J Med Genet. 2000;95:233-6.
- [Google Scholar]
- The camptodactyly-arthropathy-coxa vara-pericarditis syndrome: clinical features and genetic mapping to human chromosome 1. Arthritis Rheum. 1998;41:730-5.
- [Google Scholar]
- CACP, encoding a secreted proteoglycan, is mutated in camptodactyly-arthropathy-coxa vara- pericarditis syndrome. Nat Genet. 1999;23:319-22.
- [Google Scholar]
- A novel deletion mutation in proteoglycan-4 underlies camptodactyly-arthropathy-coxa-vara-pericarditis syndrome in a consanguineous Pakistani family. Arch Med Res. 2011;42:110-4.
- [Google Scholar]
- A novel mutation in PRG4 gene underlying camptodactyly-arthropathy-coxa vara-pericarditis syndrome with the possible expansion of the phenotype to include congenital cataract. Birth Defects Res A Clin Mol Teratol. 2012;94:553-6.
- [Google Scholar]
- CACP syndrome: identification of five novel mutations and of the first case of UPD in the largest European cohort. Eur J Hum Genet. 2014;22:197-201.
- [Google Scholar]
- Camptodactyly-arthropathy-coxa vara-pericarditis syndrome in Saudi Arabia: Clinical and molecular genetic findings in 22 patients. Semin Arthritis Rheum. 2013;43:292-6.
- [Google Scholar]
- Comparison of the boundary-lubricating ability of bovine synovial fluid, lubricin, and Healon. J Biomed Mater Res. 1998;40:414-8.
- [Google Scholar]
- The isolation and partial characterization of the major glycoprotein (LGP-I) from the articular lubricating fraction from bovine synovial fluid. Biochem J. 1977;161:473-85.
- [Google Scholar]
- The molecular structure and lubricating activity of lubricin isolated from bovine and human synovial fluids. Biochem J. 1985;225:195-201.
- [Google Scholar]
- A novel proteoglycan synthesized and secreted by chondrocytes of the superficial zone of articular cartilage. Arch Biochem Biophys. 1994;311:144-52.
- [Google Scholar]
- Immunodetection and partial cDNA sequence of the proteoglycan, superficial zone protein, synthesized by cells lining synovial joints. J Orthop Res. 1999;17:110-20.
- [Google Scholar]
- Consequences of disease-causing mutations on lubricin protein synthesis, secretion, and post-translational processing. J Biol Chem. 2005;280:31325-32.
- [Google Scholar]
- Hemangiopoietin, a novel human growth factor for the primitive cells of both hematopoietic and endothelial cell lineages. Blood. 2004;103:4449-56.
- [Google Scholar]
- The role of lubricin in the mechanical behavior of synovial fluid. Proc Natl Acad Sci USA. 2007;104:6194-9.
- [Google Scholar]
- Homology of lubricin and superficial zone protein (SZP): products of megakaryocyte stimulating factor (MSF) gene expression by human synovial fibroblasts and articular chondrocytes localized to chromosome 1q25. J Orthop Res. 2001;19:677-87.
- [Google Scholar]
- Genomic patterns of homozygosity in world-wide human populations. Am J Hum Genet. 2012;91:275-92.
- [Google Scholar]




