Translate this page into:
New treatment strategies for Alzheimer's disease: is there a hope?
Reprint requests: Dr Orestes V. Forlenza, Laboratory of Neuroscience (LIM-27) Department and Institute of Psychiatry, Faculty of Medicine, University of São Paulo, Rua Dr. Ovídio Pires de Campos 785, 05403-010 - São Paulo, SP, Brazil e-mail: forlenza@usp.br
-
Received: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Alzheimer's disease (AD) is a progressive and irreversible neurodegenerative disease, and corresponds to the most common cause of dementia worldwide. Although not fully understood, the pathophysiology of AD is largely represented by the neurotoxic events triggered by the beta-amyloid cascade and by cytoskeletal abnormalities subsequent to the hyperphosphorylation of microtubule-associated Tau protein in neurons. These processes lead respectively to the formation of neuritic plaques and neurofibrillary tangles, which are the pathological hallmarks of the disease. Clinical benefits of the available pharmacological treatment for AD with antidementia drugs (namely cholinesterase inhibitors and memantine) are unquestionable, although limited to a temporary, symptomatic support to cognitive and related functions. Over the past decade, substantial funding and research have been dedicated to the search and development of new pharmaceutical compounds with disease-modifying properties. The rationale of such approach is that by tackling key pathological processes in AD it may be possible to attenuate or even change its natural history. In the present review, we summarize the available evidence on the new therapeutic approaches that target amyloid and Tau pathology in AD, focusing on pharmaceutical compounds undergoing phase 2 and phase 3 randomized controlled trials.
Keywords
Alzheimer's disease
antidementia drug
beta-amyloid
cognitive impairment
Tau
treatment

Introduction
Alzheimer's disease (AD) is the leading cause of dementia worldwide, affecting more than half of the overall number of demented individuals, which has been estimated to be around 24 million across all nations1. The prevalence of dementia is expected to further increase in the forthcoming decades, as a consequence of the steady growth of ageing population in both developed and developing countries. According to World Health Organization projections, about three-quarters of the estimated 1.2 billion elders will be living in low- and middle-income countries by the year 20252. Age-adjusted estimates of dementia prevalence are high (above 5%) in most Asian and Latin American Countries3. However, prevalence rates of dementia seem to be lower (1-3%) in India and sub-Saharan Africa3.
Epidemiological studies conducted in India between 1996 and 2006 indicated that dementia affects 2.7% of the population, AD being the most common cause (1.3%)3. It is noteworthy that these numbers parallel the estimates of dementia and AD in Western societies cut by half; nonetheless the proportion of cases of AD amongst dementia is basically the same. Lower rates of dementia in India might be interpreted as related to environmental and biological factors such as dietary habits, lifestyle, sociocultural, cardiovascular, and genetic4. In a cross-national epidemiological study addressing the association between the apolipoprotein E genotype and the diagnosis of AD, Ganguli et al5 indicated that the frequency of the APOE*E4 allele, an important genetic risk factor for late-onset AD, was lower (7%) in northern India as compared to a North-American cohort (11%); interestingly, when the study group was subdivided according to the presence of this risk allele, the strength of association between APOE*E4 and the diagnosis AD was similar both in Indian and US samples5. Methodological reasons must also be considered as a possible source of bias in the previous estimates of dementia prevalence in India, such as non-recognition of cases due to limited access of patients to specialized services in remote regions, selective removal of cases due to early mortality, and culturally-related difficulties to ascertain the diagnosis of cognitive impairment4. A recent epidemiological study indicated that the prevalence of dementia and AD in south India (6.44 and 3.56%, respectively) was actually higher than previously reported6. Additionally, an autopsy-based study demonstrated that the prevalence of AD pathology in India was similar to that observed in developed countries7.
Studies have also suggested dissimilarities in the natural course of the disease in India as compared to western countries. As part of the Indo-US study, Chandra et al8 reported a lower median survival time after the onset of symptoms of patients with AD in India, as compared to developed countries. Potential causes of this shorter life-expectancy of AD patients are complex and may be secondary to ineffective public health assistance evolving dementia care, but other social, environmental and biological factors may be associated to a more rapid course of the illness. AD patients in India may have significantly more delusions, hallucinations, anxiety and mood symptoms during the course of dementia9. With respect to the prodromal stages of AD, a cross-sectional study in India reported a 15 per cent prevalence of mild cognitive impairment (MCI) among individuals aged 50 yr and older, multiple-domain MCI being the most prevalent subtype (8.9%)10. These findings are similar to those derived from studies performed in developed countries, and were shown to be significantly associated with increasing age, hypertension, and diabetes mellitus. Therefore, the interaction between underlying clinical risk factors and primary degenerative mechanisms in the ageing population is likely to boost the incidence of dementia in India in the near future10.
The so-called “AD-epidemic” will inevitably represent a major public health problem to most nations, because to date there is no effective approach in terms of cure or prevention of dementia. The available pharmacological therapy with antidementia drugs is largely symptomatic, with temporary clinical benefits on cognitive, functional and behavioural manifestations of the disease. This strategy relies on the increment of synaptic availability of acetylcholine, given the cholinergic deficit that arises from neuronal loss in basal forebrain nuclei and cortical projections. This effect is achieved by inhibition of acetyl-cholinesterase by donepezil, galantamine or rivastigmine. Memantine is a voltage-dependent N-methyl D-aspartate (NMDA) receptor antagonist that precludes the excessive release of glutamate as a consequence of nerve cell damage in the cortex. Therefore, the clinical use of cholinesterase inhibitors is indicated for patients with mild, moderate and even severe AD; moderate and severe cases can also be treated with memantine, preferably in combination with the former agents.
In this context, there is an urgent need to develop new drugs with disease-modifying properties for AD. By definition, a disease-modifying drug is a pharmaceutical agent intended to slow the progression of the neurodegenerative process by inhibiting critical events in the pathophysiology of the disease, and therefore, attenuating the pathological load. Allegedly, patients treated with such agents would be expected to have a more benign course of the disease when compared to placebo-treated individuals (Fig. 1). Many candidate drugs with distinct pharmacological mechanisms have been proposed in the recent years and tested in neurobiological models of AD. However, the promising effects that had been attributed to some of these compounds in animal models failed to prove beneficial to humans in early clinical trials. In addition, other drugs that underwent clinical experimentation delivered negative results, either due to limited efficacy or toxicity, when tested by randomized-controlled trials (RCTs)11. In the present review, we summarize the available results of candidate treatments for AD targeting the pathological cascades related to beta-amyloid and Tau, and focusing on pharmaceutical compounds that have been tested in phase 2 and phase 3 RCTs.
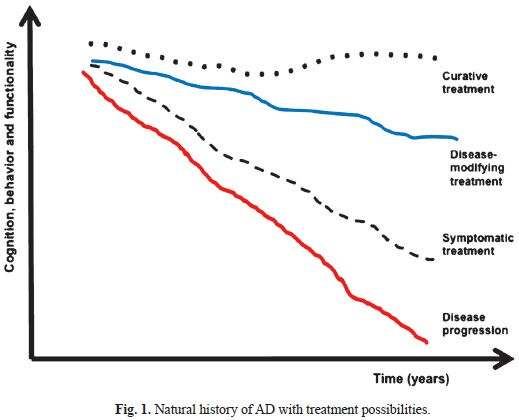
- Natural history of AD with treatment possibilities.
Pathological mechanisms in AD
The “amyloid hypothesis” is a prevailing pathological model of AD, accepted by most researchers. According to this model, the overproduction and accumulation of amyloid-beta (Aβ) peptides in plaques represent an early and central event in the pathophysiology of AD leading to the formation of neuritic plaques, whereas the cytoskeletal changes that arise from the hyperphosphorylation and intracellular aggregation of microtubule-associated protein Tau to form neurofibrillary tangles are regarded as downstream phenomena. These changes evolve in a non-linear and dynamic way, under the effect of other predisposing factors. Both pathological cascades impair neuronal homeostasis and eventually result in cell death and neurodegeneration. These changes manifest clinically with slowly progressive cognitive decline (evolving from mild cognitive impairment to dementia), behavioural symptoms and functional impairment121314. Amyloid-β is originated by the alternative (possibly abnormal) cleavage of the amyloid precursor protein (APP) into smaller peptides (Aβ1-40 and Aβ1-42), depending on the balance between the activity of major APP cleaving enzymes, i.e., α-secretase (non-amyloidogenic) and β- and γ-secretases (amyloidogenic). In AD, the abnormal regulation of APP secretases, leading to the predominant activation of β- and γ-pathways, is associated with the overproduction of toxic amyloid species. Aβ oligomers (small aggregates of 2 to 12 peptides) and amyloid plaques exert neurotoxic effects; Aβ1-42 is more prone to form insoluble aggregates (and therefore more toxic) than Aβ1-40. The interaction between genetic and environmental factors, along with the homeostatic changes that pertain to the ageing process per se, seems to affect the balance between production and clearance of toxic Aβ peptides14.
Neurofibrillary tangles (NFTs), another pathological hallmark of the disease, are largely constituted by intracellular aggregates of paired helical filaments (PHFs), which arise from the collapse of the neuronal cytoskeleton. The structure and function of microtubules are impaired as a consequence of the abnormal hyperphosphorylation of Tau protein, which precludes its ability to stabilize the monomers of alpha- and beta-tubulin14. Hyperphosphorylated Tau aggregates into oligomers to form PHFs to further originate NFTs15. Several protein kinases are involved in this process, namely glycogen synthase kinase-3 beta (GSK3β), cyclin-dependent kinase-5 (CDK5), and extracellular signal-related kinase-2 (ERK2)16; these enzymes may also be regarded as potential targets for disease-modification, upon their inhibition by specific drugs. GSK-3β, the most important Tau kinase in neurons, is overactive in AD and its overexpression has been shown to hyperphosphorylate Tau in transgenic mouse models of AD1718. The inhibition of GSK-3β not only precludes the hyperphosphorylation of Tau, but also yields the dephosphorylation of its abnormally hyperphosphorylated epitopes by the action of protein phosphatases. Therefore, the interruption and reversal of this process may restore microtubular structure and function18. Moreover, GSK-3 inhibition downregulates the amyloidogenic cleavage of APP, which provides further evidence of the cross-talk between these two major pathological cascades in AD19.
Therapeutic targets for disease modification in AD
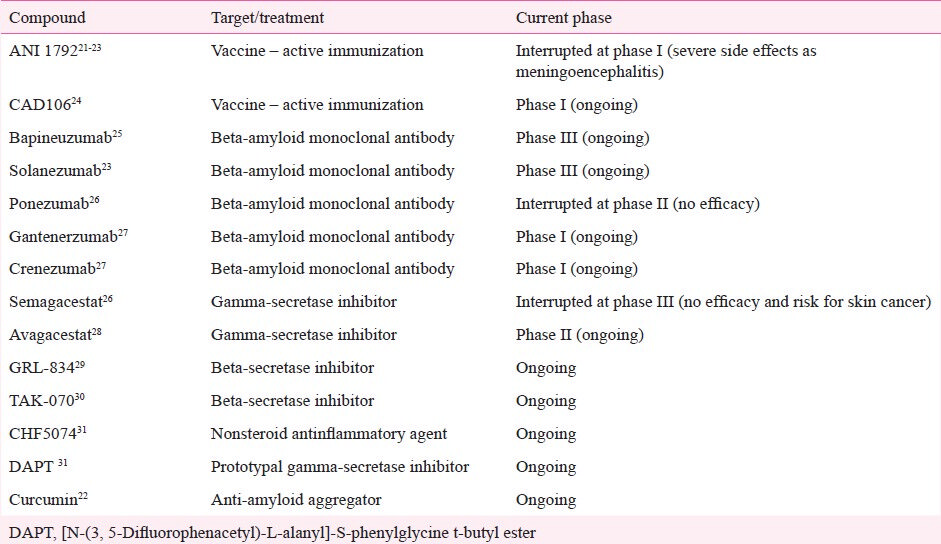
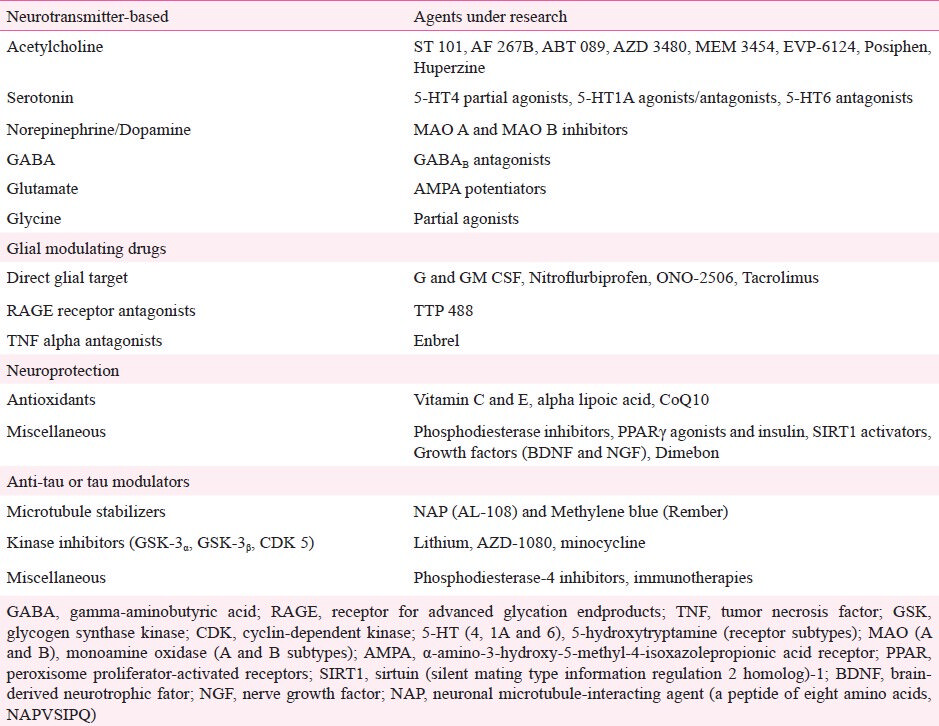
The recognition of core and secondary pathophysiological mechanisms in AD has led to the identification of molecular targets for the development of specific drugs. In fact, more than 200 pharmaceutical compounds are currently undergoing phase 2 and 3 trials11. These compounds can be grossly divided into anti-amyloid agents and drugs that target other pathological pathways. Anti-amyloid compounds can be subdivided into drugs designed to block or inhibit the overproduction or aggregation of Aβ, or to favour its clearance from the brain (Table I; Fig. 2)20, whereas the latter group can be subdivided according to predominant mechanism of action of the drug, i.e., neurotransmitter and cell-signalling agents, glial cell modulators, neuroprotective agents, and Tau-based therapies (Table II). Studies involving stem-cell and gene therapy are also under way, but at more incipient stages of experimental validation.
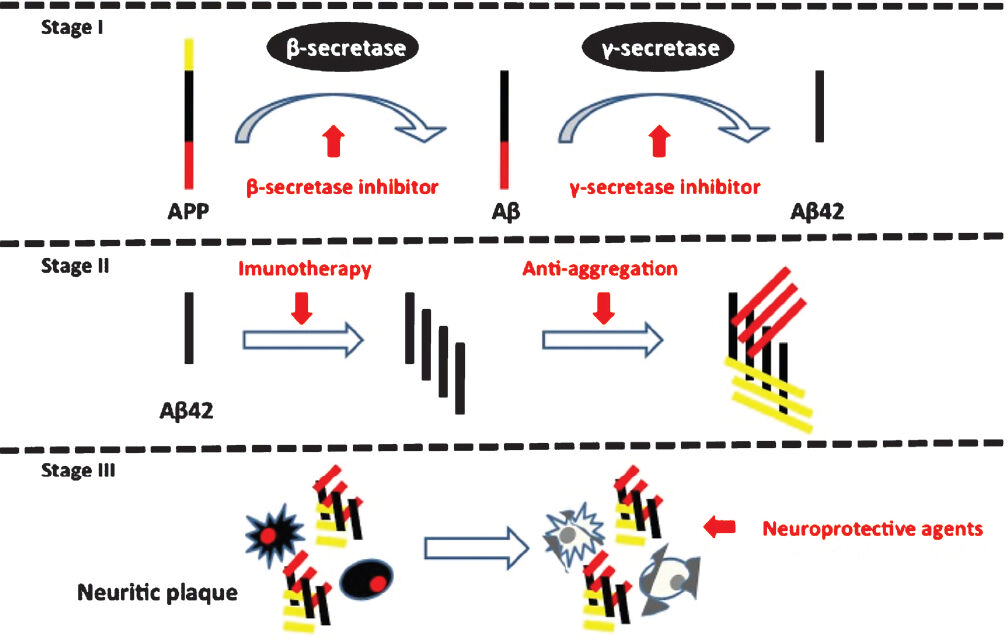
- Stages of amyloid (Aβ) beta production with possible targets for treatment. Red arrows indicate possible interventional targets with respective agent.
In AD, pathophysiological mechanisms change soluble Aβ peptides into fibrillary oligomers and insoluble fibrils, which accumulate extracellularly in the neural tissue and also in the intima of brain and systemic vessels34. Extracellular Aβ oligomers and fibrillary forms cause synaptic dysfunction, affect axons and dendritic spines, and eventually lead to neuronal loss in AD35. Toxic Aβ species also trigger secondary pathological mechanisms (e.g., oxidative stress and inflammation), which hasten neuronal dysfunction and death36. Therefore, pharmacological compounds that favour the clearance of Aβ from the brain, or prevent its aggregation, may represent a strategy to delay the progression of the pathological process in AD. Intracerebral amyloidosis may start in the brain of individuals with AD many years before the onset of clinical symptoms3738. Evidence of this pathological process can be depicted at prodromal or even at preclinical stages of the disease by the analysis of cerebrospinal fluid (CSF) and molecular neuroimaging biomarkers373839. Concentrations of soluble Aβ in the CSF, which are known to be reduced in patients with MCI who convert to dementia, have been inversely correlated to the rate of cognitive decline and to the severity of neurodegeneration3940. Although molecular imaging with PET provides measurement of pathological deposition of extracellular fibrillar Aβ, which brings plaques into the brain, there is no definitive neuroimaging confirmation for synaptic dysfunction with currently available tracers41.
Anti-amyloid therapy
Anti-amyloid strategies comprise pharmaceutical compounds with distinct mechanisms of action, namely drugs that (i) facilitate the clearance; (ii) inhibit the production; or (iii) prevent the aggregation of Aβ32. As shown in Table I, many pharmacological compounds have been developed to tackle the “amyloid cascade”, with the prospect of reducing the Aβ burden in the brain of mild to moderately demented AD patients2042. However, there is evidence that these interventions must be implemented at earlier stages of the disease process, i.e., at the stage of incipient dementia or prior to the conversion from mild cognitive impairment to dementia (in the MCI-AD continuum), or even earlier. It is accepted that two thirds of individuals with MCI may have intracerebral amyloid burden comparable with those with clinically manifested AD, indicating that intracerebral amyloidosis is an early event in the natural history of the disease4344.
Both active and passive immunization target the reduction of intracerebral Aβ load by eliciting humoral response against the Aβ peptide, facilitating its clearance from the brain by immune-mediated mechanisms45. Highly encouraging findings were presented by preclinical studies with transgenic mice with high Aβ load, submitted to active and passive immunization; these strategies proved effective reducing the amount of Aβ in the mouse brain, which was supposedly associated with improvements on behaviour and cognition4647. These studies have provided the rationale for the first generation of immunotherapeutic agents for AD, which was based on the active immunization of AD patients with the actual Aβ peptide48. Therefore, this strategy induces an IgM response to generate antibodies against pathogenic Aβ, which further mobilize microglia to clean plaques through phagocytosis3245. Also, the immune response prevents Aβ deposition by removing the excess of soluble Aβ forms from the circulation45. Phase 2A studies with the active anti-Aβ vaccine (AN 1792) proved efficacious to remove Aβ plaques from the brain of AD patients; however, the trial was interrupted due to the occurrence of severe adverse events in a substantial proportion of subjects (6%) who received this intervention, which was associated with the induction of a cytotoxic T-cell reaction in brain vessels leading to acute meningoencephalitis21. Interim analyses indicated that although the immunization promoted Aβ clearance, it was not associated with clinically relevant benefits, i.e., the immunological response was not associated with the successfully completed clinical trials2223.
The second-generation of active anti-Aβ immunotherapeutic agents was designed to minimize the risk of eliciting such secondary inflammatory responses or vasogenic oedema, by stimulating soluble Aβ derivative immunogens. These vaccines elicit the immune response to raise antibodies against Aβ monomers and oligomers. Studies with the vaccine CAD106 at phase 1 indicated that it was able to reduce Aβ accumulation in cortical and subcortical brain regions by binding to Aβ aggregates and blocking cellular toxicity, with no evidence of microhaemorrhage, vasogenic oedema, or inflammatory reactions subsequent to activation of T-cells24.
Conversely, passive immunotherapy is based on the intravenous administration of full monoclonal antibodies or antibody fragments from specific exogenous origins, which directly target Aβ34. This strategy is supposedly not associated with a significant risk of eliciting T-cell inflammatory reactions45. In passive immunization, the binding of antibodies to specific Aβ epitopes induces plaque clearance through microglial activation48. In transgenic mice, peripheral infusion of antibodies raised specifically against the Aβ peptide was shown to reduce brain amyloid load27. As occurred with active immunotherapy, successful preclinical studies with passive immunotherapy were followed by phase 1 and phase 2 trials for AD. Afterward, advantageous results with transgenic animal models encouraged several centers to design clinical trials in order to establish effective treatments for AD patients and to change the disease course. Currently, some passive immunotherapies are being developed. A challengeable aspect concerns the efficacy against pathophysiology of the disease with avoidance of undesired side effects such as microhaemorrhage, vasogenic oedema, encephalitis, or neuroimaging abnormalities27. Across the screening cohorts enrolling placebo-treated patients, rarely occurrences of brain areas of microhaemorrhage have been described4950. Conversely, another group reported vasogenic oedema with this drug28.
Several passive immunotherapeutic agents have been evaluated by RCTs over the past years, namely bapineuzumab, solanezumab, gantenerumab, ponezumab, and crenezumab. These monoclonal antibodies have high affinity to antigenic determinant epitopes of Aβ, binding either to soluble forms or in plaques, being further recognized by B- and T-cells to promote its clearance from the brain2748. In addition, monoclonal antibodies may delay amyloid-β burden or stop its accumulation in the brain2748. Bapineuzumab represents the humanized Aβ monoclonal antibody that has been most tested as a candidate drug for the treatment of AD, and is currently undergoing phase 3 studies. In phase 2 studies, this compound was examined in a randomized placebo controlled trial to determine dose, safety, and efficacy in 234 patients with mild to moderate AD. Patients were given six infusions, in 13 distinct weeks, with last evaluation at week 7825. Ascending doses of bapineuzumab were shown to reduce intracerebral Aβ load through increased clearance from the brain; however, this effect was not associated with a significant benefit in clinical outcomes. Reversible and asymptomatic or transient vasogenic oedema was detected in 9.7 per cent of immunized patients, being more frequent in those receiving higher doses and in APOE*4 carriers25. Recently, during The European Federation of Neurological Societies annual meeting, in Stockholm, Sweden (2012), researchers reported that bapineuzumab failed to protect against cognitive and functional decline of AD patients undergoing a phase 3 trial23.
Solanezumab is another humanized monoclonal antibody that binds to the mid domain of soluble forms of Aβ peptide. Preliminary results indicated that it was effective promoting Aβ clearance from the brain; however, two placebo-controlled phase 3 trials failed to demonstrate clinical benefits23. Rare occurrences of micro-haemorrhage and vasogenic oedema have also been described with this drug2849. In a study conducted in Japan with AD patients with mild to moderate dementia, treatment with solanezumab was associated with a significant increase in plasma concentrations of the Aβ peptide, reflecting the increased clearance of Aβ from the brain, in the absence of relevant side effects50.
The monoclonal antibody ponezumab targets the amino-terminal portion of Aβ1-40. In animal models this antibody significantly reduced cerebral Aβ burden and cerebral amyloid angiopathy, along with improvements in behaviour27. In a phase 1 study, a single intravenous dose of ponezumab was shown to be safe and well tolerated, and associated with increments in plasma and CSF concentrations of the Aβ peptide, suggesting that the drug may alter central Aβ levels. However, subsequent phase 2 studies did not confirm clinical efficacy, and development of ponezumab for mild to moderate AD was interrupted26. There are other ongoing trials with humanized monoclonal antibodies raised against the Aβ peptide, such as gantenerumab and crenezumab. Gantenerumab seems to reduce the cerebral Aβ load in AD patients in a dose-dependent fashion27. However, it is yet to be determined whether treatment with this antibody can slow disease progression and improve clinical outcomes49. Data from a phase 1 clinical trial testing another humanized anti-Aβ monoclonal antibody, known as MABT5102A, to protect against Aβ1-42 oligomer-induced cytotoxicity, showed no vasogenic oedema, even among APOE*4 carriers50.
Other anti-amyloid strategies have been addressed by clinical trials. Preliminary studies support that the production and accumulation of Aβ can be downregulated by the specific γ-secretase inhibitors avagacestat and semagacestat2628.
Recently, semagacestat, a non-selective gamma-secretase inhibitor, has been examined as a potential treatment for AD patients51. Unfortunately, preliminary results from two ongoing long-term phase III trials found no efficacy. The studies were interrupted because researchers verified it did not slow disease progression and, in addition, was associated with increasing cognitive impairment and worsening daily living activities, as well with increasing risk for skin cancer with semagacestat51. Although vasogenic oedema has been a rare vascular event, a report described exacerbation of psoriatic skin lesions with this drug2152.
Avagacestat has been considered as a potently inhibitor of Ab40 and Ab42 formation, with selectivity for effects on APP relative to Notch proteins which interfere with cell proliferation, differentiation, and apoptosis. In a study enrolling healthy subjects, this compound exerted a potent selective gamma-secretase inhibition with decreased CSF Ab levels, as well as the inhibition of the human Notch proteins53. The authors reported a good tolerability profile and no changes in histopathology of skin or in lymphocytes following 28 days of dosing, although this period was not enough to confirm such clinical alterations in humans52. A phase II study with AD outpatients reported a dose-dependent of avagacestat on CSF amyloid isoforms and Tau protein. However, the authors demonstrated no signicant reduction in these biomarkers, although they reported an acceptable safety and tolerability with this drug54. Another study was designed to compare chronic treatment of transgenic mice models of AD between the nonsteroidal anti-inflammatory CHF5074 and a prototypal gamma-secretase inhibitor (DAPT)31. The authors observed that the CHF5074 compound has effectively prevented amyloid accumulation in the brain tissue and behavioural impairment; however, no significant effects were found with DAPT treatment.
The inhibition of beta-secretase is another potential mechanism of disease modification in AD, given the major role of this enzyme in the amyloidogenic cleavage of the amyloid precursor protein (APP). BACE-1 (β-site amyloid precursor protein cleaving enzyme 1) produces two peptides (Ab40 and Ab42), and its inhibition with specific compounds precludes the excess of amyloid and its accumulation into plaques55.
An inhibitor of β-secretase (GRL-8234) was recently investigated in young transgenic mice with decreased soluble amyloid-beta in the brain tissue and with rescued behaviour performance29. These findings indicated that β-secretase inhibitors play a role in reduction of amyloid-beta plaque load at an early stage of AD pathogenesis, as well as in older mice, suggesting possible benefits for treatment of AD patients at a later stage of the disease29.
TAK-070 is a non-petpidic agent that inhibits BACE-1 in a dose-dependent and non-competitive maner. However, the inhibitory mechanism of this compound are not clear, and the reduction of amyloid-beta has been considered modest with low selectivity over other enzymes30. Using an AD transgenic mouse model, the immunogenicity and efficacy of the compound AF20513 was tested and it was found that this active vaccine induced cellular and humoral immune responses against beta-amyloid pathophysiology in the brain tissue of the animals, without inducing microglial activation56. In addition, the amyloid-β anti-aggregator curcumin could be a future therapeutic strategy22. However, large clinical trials are needed to test this and other promising drugs.
The occurrence of vasogenic oedema and multiple cortical and subcortical vascular lesions represent adverse events that must be considered in Aβ immunotherapy with monoclonal antibodies. These radiological findings are associated with cerebral amyloid angiopathy-related inflammation (CAA-ri), and can be asymptomatic or present clinically with acute or sub-acute manifestations such as headache, seizures, focal neurological deficits, and behavioural disturbances5758. This reaction was observed with bapineuzumab and other monoclonal antibodies but, interestingly, it was also reported in patients treated with the γ-secretase inhibitor avagacestat2659.
Tau-oriented strategies
Given its critical role in pathogenesis of AD, drug development may also target the production, processing (phosphorylation) and aggregation of Tau protein33. Three ‘anti-tau’ strategies have been addressed by clinical trials. Agents like methylene-blue (Rember) and NAP (AL-108) favour the stabilization of microtubules, while lithium salts may prevent Tau hyperphosphorylation through the inhibition of GSK-3β60. Two widely used mood stabilizers, lithium and valproate, are also inhibitors of this enzyme, reducing tau phosphorylation in animal models61. Proteomic studies in AD patients treated with valproate indicated ten differentially expressed proteins related to functional classes implicated in the neurobiology of the disease and to the therapeutic action of drug62. However, in a multicenter clinical trial conducted by the Alzheimer's Disease Cooperative Study (ADCS), an accelerated decrease in total brain and hippocampal volume was observed after 1 year of followup among patients treated with divalproex sodium, which was accompanied by greater cognitive impairment63. In addition, valproate treatment of moderately demented AD patients did not delay the emergence of neuropsychiatric symptoms of agitation or psychosis, and was not associated with reduction of cognitive or functional decline; rather, it was associated with adverse effects such as somnolence, gait disturbance, tremor, diarrhoea and weakness64.
Lithium, a potent GSK-3 inhibitor, has shown neuroprotective action such as reducing Tau concentration and phosphorylation, via the inhibition of GSK-3β, reducing Aβ production, axonal degeneration and releasing the transforming growing factor beta 1 (TGF-β1) in experimental studies of cultured neurons and transgenic mice656667686970. Epidemiologic studies showed a reduced risk of developing dementia among patients with bipolar disorder treated chronically with lithium7172, but there is limited evidence of the clinical benefits of lithium treatment for patients with or at risk for AD73747576. In a recent single-centre, placebo-controlled trial conducted in a sample of patients with amnestic MCI, long-term lithium treatment was associated with stabilization of cognitive and functional parameters, in addition to a reduction in CSF concentrations of phosphorylated Tau76. In a previous phase 2 study in patients with mild AD treated with lithium for a shorter (10 wk) period, no significant differences were observed in biological (GSK-3 activity and CSF biomarkers) or clinical (cognitive and functional) outcomes74. In addition to that, the safety limits of chronic lithium treatment for older adults need to be determined, given the risk of renal impairment, hypothyroidism and other metabolic adverse effects77. Other pharmaceutical compounds targeting GSK-3β, such as the specific inhibitors AR-A014418, SRN-003-55, CHIR-98014 and SB216763, have so far shown neuroprotective effects in preclinical models of AD78.
Methylthioninium chloride (known as Rember), also known as methylene-blue, is a phenothiazine widely used as a histological stain. Currently, it is a promising compound being evaluated in AD trials. The drug has anti-oxidative properties and has been shown to reduce Aβ oligomerization, given its ability to bind to the Tau domain that is required for its aggregation, therefore, preventing the formation of PHFs79. In animal models, it has been shown to decrease Aβ levels and prevent cognitive deficits in triple transgenic mice80. A phase 2b randomized trial of Rember monotherapy in mild and moderate AD patients suggested cognitive benefits in volunteers81. Results of a phase 3 trial are soon expected to assure its safety profile and efficacy as a disease-modifying drug for AD82.
Conclusions and future directions
To date, treatment of AD relies on the symptomatic effects of cholinesterase inhibitors and NMDA-receptor antagonists. Many promising compounds have been validated by experimental models as candidate disease-modifying drugs for AD; however, only a few of these appear in the pipeline of drug development, or have been clinically tested by RCTs. Overall results from these trials have so far been negative. Most phase 3 trials with candidate drugs for AD in the last decade failed to present unequivocal clinical benefits, or were suspended due to severe adverse events. Likewise, results of ongoing phase 2 trials are discrete at most. This rather pessimistic scenario must be weighed against the huge financial costs of the transition from phase 2 to phase 3 trials, and further translation of this knowledge into clinical practice. Several issues must be addressed in future studies with candidate drugs in order to yield clinically relevant results, and support their generalization for the treatment of the massive population of patients with (and at risk for) AD. One critical issue refers to the stage of the disease in which one given treatment is prone to deliver clinically relevant benefits. The majority of studies with candidate disease-modifying drugs for AD recruited patients with clinically manifested dementia, when it may be too late to grant clinical benefits from the modification of the pathological process. In other words, by treating demented patients with such drugs it may be possible to prove the concept of disease modification, but this effect may not be translated into significant changes to the natural course of the disease. Forthcoming trials should rather target disease modification in patients with very mild clinical symptoms (such as incipient dementia or amnestic MCI), i.e., at prodromal or even pre-clinical stages of AD83. A recent consensus paper published by the National Institute of Aging/Alzheimer's Association working group proposed a three-stage stratification of patients that could be used to guide for the recruitment populations for secondary prevention or interventional trials84. With respect to trials in MCI, it is essential to improve the definition of cases, i.e., to recruit amnestic MCI patients with true AD pathology and, therefore, higher risk of conversion to dementia. For this purpose, recruitment of patients for future studies should be rationally based on new diagnostic technologies including biochemical and imaging biomarkers (e.g., CSF concentrations of Aβ42, total Tau and phosphor-Tau, molecular imaging of amyloid with PET). Another important point refers to the duration of trials, and the best moment to start the intervention, given the long period of time prior to the onset of symptoms in which the pathological changes are evolving within the brain. In other words, it is imperative to develop a better understanding of the disease process and the possibilities of modification of the AD pathogenesis, which requires evidence-based answers for the following three questions: who should be treated; when to start; and for how long? Finally, one should better define the outcomes of these trials, with biological measures to prove that the drug is efficacious in disease-modification even in the absence of significant clinical improvement. Therefore, the notion of prevention of dementia in at-risk individuals must be reinforced.
References
- World Health Organization (WHO). Active ageing: a policy framework. Geneva: WHO; 2002. WHO/NMH/NPH/028
- [Google Scholar]
- Alzheimer's disease and vascular dementia in developing countries: prevalence, management, and risk factors. Lancet Neurol. 2008;7:812-26.
- [Google Scholar]
- Risk factors of dementia in North India: a case-control study. Aging Ment Health. 2012;16:228-35.
- [Google Scholar]
- Apolipoprotein E polymorphism and Alzheimer disease: the Indo-US Cross-National Dementia Study. Arch Neurol. 2000;57:824-30.
- [Google Scholar]
- Dementia in Kerala, South India: prevalence and influence of age, education and gender. Int J Geriatr Psychiatry. 2010;25:290-7.
- [Google Scholar]
- Profiles of Alzheimer's disease-related pathology in an aging urban population sample in India. J Alzheimers Dis. 2011;24:187-96.
- [Google Scholar]
- Prevalence of Alzheimer's disease and other dementias in rural India: the Indo-US study. Neurology. 1998;51:1000-8.
- [Google Scholar]
- Behavioral and psychological symptoms of dementia in an Indian population: comparison between Alzheimer's disease and vascular dementia. Int Psychogeriatr. 2006;18:87-93.
- [Google Scholar]
- An epidemiologic study of mild cognitive impairment in Kolkata, India. Neurology. 2007;68:2019-26.
- [Google Scholar]
- Current insights into the pathophysiology of Alzheimer's disease: selecting targets for early therapeutic intervention. Int Psychogeriatr. 2012;24(Suppl 1):S10-7.
- [Google Scholar]
- Has the amyloid cascade hypothesis for Alzheimer's disease been proved? Curr Alzheimer Res. 2006;3:71-3.
- [Google Scholar]
- The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem. 2009;110:1129-34.
- [Google Scholar]
- Tau oligomers and aggregation in Alzheimer's disease. J Neurochem. 2010;112:1353-67.
- [Google Scholar]
- Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol. 1999;58:1010-9.
- [Google Scholar]
- GSK-3alpha regulates production of Alzheimer ìs disease amyloid -beta peptides. Nature. 2003;423:435-9.
- [Google Scholar]
- The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science. 2002;297:353-6.
- [Google Scholar]
- Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448-52.
- [Google Scholar]
- Multitargeted drugs discovery: balancing anti-amyloid and anticholinesterase capacity in a single chemical entity. Bioorg Med Chem Lett. 2011;21:2655-8.
- [Google Scholar]
- The European Federation of Neurological Societies annual meeting, in Stokholm. 2012. Available from: http://www.efns.org
- [Google Scholar]
- The second-generation active Aβ immunotherapy CAD106 reduces amyloid accumulation in APP transgenic mice while minimizing potential side effects. J Neurosci. 2011;31:9323-31.
- [Google Scholar]
- A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73:2061-70.
- [Google Scholar]
- Safety and pharmacology of a single intravenous dose of ponezumab in subjects with mild-to-moderate Alzheimer disease: a phase I, randomized, placebo-controlled, double-blind, dose-escalation study. Clin Neuropharmacol. 2013;36:14-23.
- [Google Scholar]
- Safety and pharmacology of ponezumab (PF-04360365) after a single 10-minute intravenous infusion in subjects with mild to moderate Alzheimer disease. Clin Neuropharmacol. 2013;36:8-13.
- [Google Scholar]
- Vasogenic edema in the setting of A-amyloid lowering therapy, adverse event: what is it and how is it detected? Alzheimers Dement. 2011;7(Suppl):e75.
- [Google Scholar]
- Beta-secretase inhibitor GRL-8234 rescues age-related cognitive decline in APP transgenic mice. FASEB J. 2011;25:775-84.
- [Google Scholar]
- Advances in the identification of β-secretase inhibitors. Expert Opin Drug Discov. 2013;8:709-31.
- [Google Scholar]
- Multi-target action of the novel anti-Alzheimer compound CHF5074: in vivo study of long term treatment in Tg2576 mice. BMC Neurosci. 2013;14:44.
- [Google Scholar]
- New pharmacological strategies for treatment of Alzheimer's disease: focus on disease modifying drugs. Br J Clin Pharmacol. 2012;73:504-17.
- [Google Scholar]
- Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112-9.
- [Google Scholar]
- Immunotherapeutic strategies for Alzheimer's disease treatment. Scientific World J. 2009;9:909-19.
- [Google Scholar]
- Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282-8.
- [Google Scholar]
- Curcumin loaded-PLGA nanoparticles conjugated with Tet-1 peptide for potential use in Alzheimers disease. PLoS One. 2012;7:e32616.
- [Google Scholar]
- Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207-16.
- [Google Scholar]
- Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging - Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280-92.
- [Google Scholar]
- Diagnosis and biomarkers of predementia in Alzheimer's disease. BMC Med. 2010;8:89.
- [Google Scholar]
- Soluble pool of A beta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860-6.
- [Google Scholar]
- Molecular imaging in Alzheimer's disease: new perspectives on biomarkers for early diagnosis and drug development. Alzheimers Res Ther. 2011;3:1-9.
- [Google Scholar]
- Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545-55.
- [Google Scholar]
- Alzheimer's Disease Neuroimaging Initiative. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355-65.
- [Google Scholar]
- Beta-amyloid burden in the temporal neocortex is related to hippocampal atrophy in elderly subjects without dementia. Neurology. 2010;74:121-7.
- [Google Scholar]
- Anti-β-amyloid immunotherapy for Alzheimer's disease: focus on bapineuzumab. Curr Alzheimer Res. 2011;8:808-17.
- [Google Scholar]
- Immunization with amyloid-beta attenuates Alzheimer disease-like pathology in the PDAPP mouse. Nature. 1999;400:173-7.
- [Google Scholar]
- A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982-5.
- [Google Scholar]
- Sustained levels of antibodies against Aβ in amyloid-rich regions of the CNS following intravenous dosing in human APP transgenic mice. Exp Neurol. 2012;238:38-43.
- [Google Scholar]
- Prevalence of asymptomatic vasogenic edema in pretreatment Alzheimer's disease study cohorts from phase 3 trials of semagacestat and solanezumab. Alzheimers Dement. 2011;7:396-401.
- [Google Scholar]
- Comparison of pharmacokinetics, pharmacodynamics, safety, and tolerability of the amyloid β monoclonal antibody solanezumab in Japanese and White patients with mild to moderate Alzheimer disease. Clin Neuropharmacol. 2012;35:25-9.
- [Google Scholar]
- 2010. Eli Lilly Company; Available from: http://newsroom.lilly.com/reliasedetail.cfm?releaseid=499794
- Exacerbation of psoriatic skin lesions in a patient with Alzheimer disease receiving gamma-secretase inhibitor. J Am Acad Dermatol. 2013;68:e46-8.
- [Google Scholar]
- Pharmacodynamics of selective inhibition of γ-secretase by avagacestat. J Pharmacol Exp Ther. 2013;344:686-95.
- [Google Scholar]
- Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch Neurol. 2012;69:1430-40.
- [Google Scholar]
- Disease modifying drugs targeting β-amyloid. Am J Alzheimers Dis Other Demen. 2012;27:296-300.
- [Google Scholar]
- Immunogenicity, efficacy, safety, and mechanism of action of epitope vaccine (Lu AF20513) for Alzheimer's disease: prelude to a clinical trial. J Neurosci. 2013;33:4923-34.
- [Google Scholar]
- Anti-amyloid β autoantibodies in cerebral amyloid angiopathy-related inflammation: implications for amyloid-modifying therapies. Ann Neurol. 2013;73:449-58.
- [Google Scholar]
- Inflammatory cerebral moyloid angiopathy and amyloid-modifying therapies: variations on the same ARIA? Ann Neurol. 2013;73:439-41.
- [Google Scholar]
- Amyloid-related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11:241-9.
- [Google Scholar]
- Activation of tau protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25-35) enhances phosphorylation of tau in hippocampal neurons. Neurosci Res. 1998;31:317-23.
- [Google Scholar]
- Can lithium or valproate untie tangles in Alzheimer's disease? J Clin Psychiatry. 2009;70:919-21.
- [Google Scholar]
- Proteomic analysis of peripheral leukocytes in Alzheimer's disease patients treated with divalproex sodium. Neurobiol Aging. 2008;29:1631-43.
- [Google Scholar]
- Chronic divalproex sodium use and brain atrophy in Alzheimer disease. Neurology. 2011;77:1263-71.
- [Google Scholar]
- Chronic divalproex sodium to attenuate agitation and clinical progression of Alzheimer disease. Arch Gen Psychiatry. 2011;68:853-61.
- [Google Scholar]
- Biomarkers in Alzheimer's disease drug development. Alzheimers Dement. 2011;7:e13-44.
- [Google Scholar]
- Lithium reduces tau phosphorylation but not A beta or working memory deficits in a transgenic model with both plaques and tangles. Am J Pathol. 2007;170:1669-75.
- [Google Scholar]
- Inhibition of glycogen synthase kinase-3beta downregulates total tau proteins in cultured neurons and its reversal by the blockade of protein phosphatase-2A. Brain Res. 2009;1252:66-75.
- [Google Scholar]
- Lithium: potential therapeutics against acute brain injuries and chronic neurodegenerative diseases. J Pharmacol Sci. 2005;99:307-21.
- [Google Scholar]
- Psychopharmacological neuroprotection in neurodegenerative disease: assessing the preclinical data. J Neuropsychiatry Clin Neurosci. 2010;22:8-18.
- [Google Scholar]
- TGF-beta1 pathway as a new target for neuroprotection in Alzheimer's disease. CNS Neurosci Ther. 2011;17:237-49.
- [Google Scholar]
- Lithium and risk for Alzheimer's disease in elderly patients with bipolar disorder. Br J Psychiatry. 2007;190:359-60.
- [Google Scholar]
- Lithium may be useful in the prevention of Alzheimer's disease in individuals at risk of presenile familial Alzheimer's disease. Med Hypotheses. 2007;71:948-51.
- [Google Scholar]
- Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 2010;9:702-16.
- [Google Scholar]
- Lithium trial in Alzheimer's disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry. 2009;70:922-31.
- [Google Scholar]
- Disease-modifying trials in Alzheimer's disease: a European task force consensus. Lancet Neurol. 2007;6:56-62.
- [Google Scholar]
- Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198:351-6.
- [Google Scholar]
- A feasibility and tolerability study of lithium in Alzheimer's disease. Int J Geriatr Psychiatry. 2008;23:704-11.
- [Google Scholar]
- A novel GSK-3beta inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35:359-67.
- [Google Scholar]
- Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci USA. 1996;93:11213-8.
- [Google Scholar]
- Methylene blue reduces A beta levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011;21:140-9.
- [Google Scholar]
- Challenges in the conduct of disease-modifying trials in AD: practical experience from a phase 2 trial of Tau-aggregation inhibitor therapy. J Nutr Health Aging. 2009;13:367-9.
- [Google Scholar]
- Revising the definition of Alzheimer's disease: a new lexicon. Lancet Neurology. 2010;9:1118-27.
- [Google Scholar]
- Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgoup. Alzheimer's Dement. 2011;7:367-85.
- [Google Scholar]




