
Translate this page into:
Molecular mechanisms of zika virus pathogenesis: An update
For correspondence: Dr Pankaj Seth, Department of Cellular & Molecular Neuroscience, National Brain Research Centre, Manesar 122 052, Gurgaon, India e-mail: pseth.nbrc@gov.in
-
Received: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Zika virus (ZIKV), member of the family Flaviviridae belonging to genus Flavivirus, is an arthropod-borne virus. The ZIKV is known to cause severe congenital birth defects in neonates. Due to a large number of worldwide outbreaks and associated neurological complications with ZIKV, a public health emergency was declared by the World Health Organization on February 1, 2016. The virus exhibits neurotropism and has a specific propensity towards neural precursor cells of the developing brain. In utero ZIKV infection causes massive cell death in the developing brain resulting in various motor and cognitive disabilities in newborns. The virus modulates cell machinery at several levels to replicate itself and inhibits toll like receptors-3 signalling, deregulates microRNA circuitry and induces a chronic inflammatory response in affected cells. Several significant advances have been made to understand the mechanisms of neuropathogenesis, its prevention and treatment. The current review provides an update on cellular and molecular mechanisms of ZIKV-induced alterations in the function of various brain cells.
Keywords
Brain development
cell cycle
flavivirus
Guillain–Barré
syndrome
microcephaly
neural stem cells
zika virus

Zika virus (ZIKV), member of the virus family Flaviviridae, genus Flavivirus, is an arthropod-borne virus. Globally, 84 countries have reported active ZIKV transmission1. Among other flaviviruses, only ZIKV is known to have teratogenic effects in humans that culminate into abnormally smaller size of head circumference, referred to as microcephaly and intracranial calcification and often foetal death2. A serological survey suggested that ZIKV was endemic to East, Central, West and South Africa and as well as in several Asian countries3. ZIKV was first isolated in 1947 from a sentinel rhesus monkey from Zika forest of Uganda in a study supported by Rockefeller foundation3.
ZIKV is a single-stranded positive-sense RNA virus with a genome size of 10.8 Kb. The genome has a single ~10 kb of open reading frame (ORF) with ~100 and ~420 nt of 5′ untranslated region (UTR) and 3′ UTR, respectively4. The ORF of ZIKV genome encodes for a single polyprotein which is later on processed into the structural and non-structural proteins of the virus. Three structural proteins are the precursor membrane (prM), the envelope protein (E), capsid (C) and seven non-structural proteins (NS) are NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5. The structural proteins PrM, C and E form the viral particle, while the seven non-structural proteins are critical for crucial viral functions during genome replication, polyprotein processing and manipulation of host responses for viral advantage5. The Figure 1 shows the genetic organization of ZIKV. The C protein makes the viral icosahedral capsid, which is surrounded by a spherical lipid bilayer membrane from the host. The M and E proteins on the viral surface form transmembrane helices which help in anchoring them onto the outer membrane of the host cell. The E protein is the principal surface viral protein involved in host cell binding and membrane fusion6.
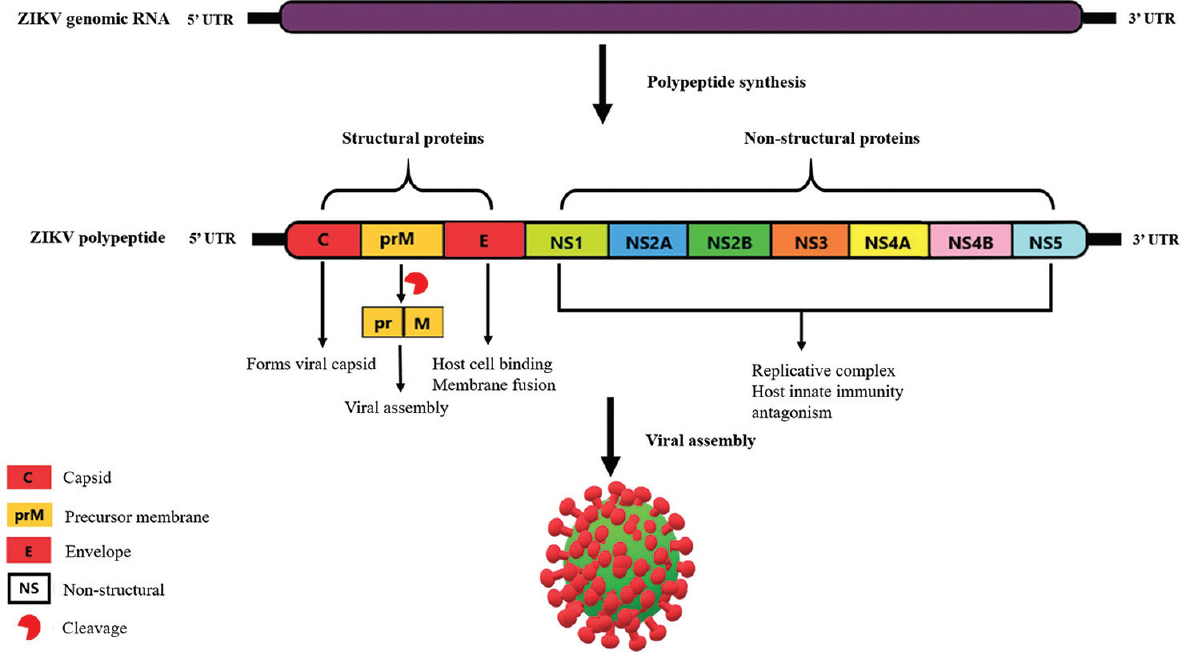
- Genetic organization of Zika virus.
ZIKV replication is similar to other flaviviruses6. The flaviviruses enter the host cell by binding to the cell surface, followed by clathrin-mediated endocytosis specifically through viral glycoprotein E-dependent manner6. Mature virus released into the extracellular space follows the conventional protein secretory pathway but the precise mechanisms of viral exit are yet to be determined. The review consolidates some of the recent advances made to understand the molecular and cellular mechanisms of ZIKV associated neuropathogenesis and highlights open questions concerning the neuropathogenesis.
Infection of zika virus
The principal mode of ZIKV transmission is through mosquito bite particularly by Aedes aegypti and Aedes albopictus. ZIKV transmission can occur through unprotected intercourse and vertical transmission has been observed from affected mother to child during pregnancy, parturition and breastfeeding. ZIKV has been detected in various body fluids such as breast milk, urine, secretions of female genital tract, amniotic fluid, saliva, cerebrospinal fluid, aqueous humour, conjunctival fluid and nasopharyngeal area7891011121314; hence, transmission can occur by exchange of any body fluids between affected and unaffected individuals. The Figure 2 shows vertical transmission of ZIKV. During initial 2-7 days of the incubation time, infected human patients serve as a source of ZIKV with low viral load and may transmit the virus through body fluids15.
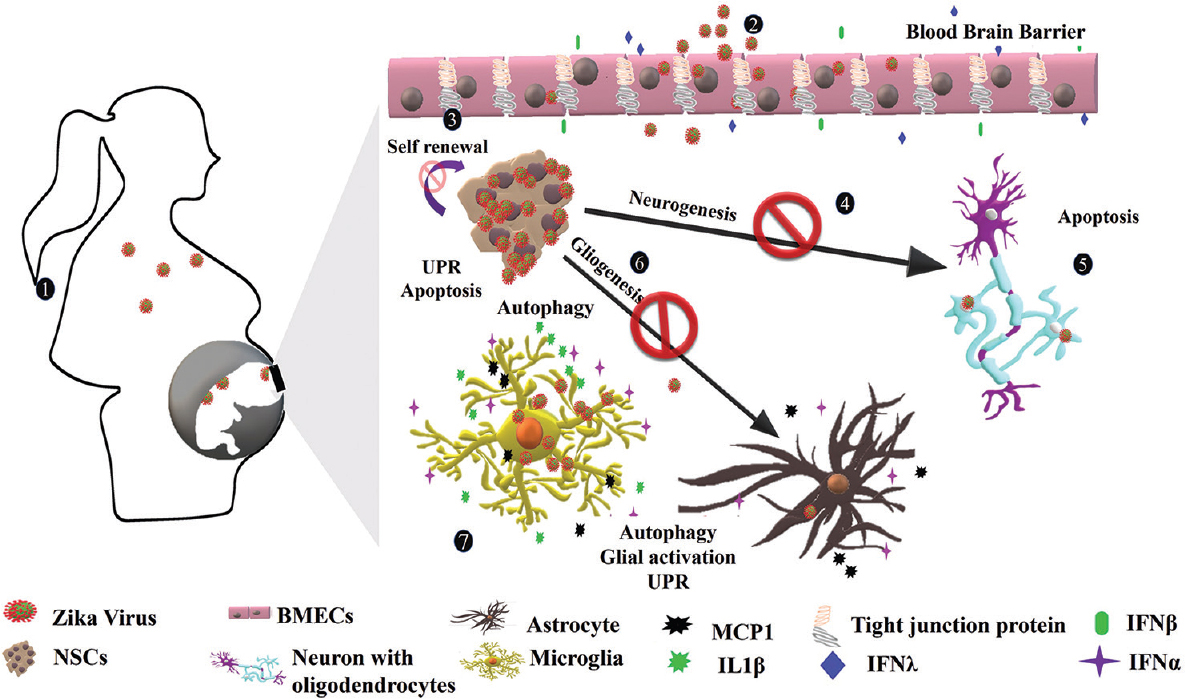
- Vertical transmission and congenital Zika Syndrome: (1) ZIKV is spread from bite of mosquitoes and potentially crosses placental barrier and infects developing brain cells of the foetus. (2) ZIKV infects BMECs that form the blood–brain barrier. The BMECs release interferon β (IFNβ), IFN λ and other interferon-stimulated genes leading to the disruption of blood–brain barrier. (3) The virus infects NSCs, the primary cell type in the developing brain and induce apoptosis, UPR and alter self-renewal of NSCs. (4) Infected NSCs have altered neurogenesis and differentiating NSCs enter apoptosis in the presence of ZIKV or its proteins. (5) Zika virus also infects oligodendrocytes and mature neurons but with low propensity. (6) Infected astrocytes release MCP1 and IFN α in response to ZIKV. The virus also induces UPR and autophagy in astrocytes. (7) Microglial cells infected with ZIKV respond robustly to release various cytokines and chemokines such as IFN α, IL-6, MCP1, TNFα, IL-1β, IL 8, MIP-1α and inducible nitric oxide synthase (iNOS). The release of various inflammatory and proinflammatory molecules from astrocytes and ZIKV suggests a severe inflammatory scenario in the developing brain.
Flaviviruses deposit on skin epidermis after the mosquito bite where they encounter cells such as keratinocytes and Langerhans cells, which are highly permissive to virus. It has been reported that the entry of the virus is through the interaction of E protein with TAM (Tyro3, Axl, and Mer), TIM (T cell/transmembrane, immunoglobin, and mucin), AXL (AXL receptor tyrosine kinase) or DC-SIGN (Dendritic cell-specific intracellular adhesion molecule-3-G rabbing N on- integrin) receptors16. Dendritic cells are the first cells targeted by flaviviruses. Once infected, these migrate to different lymphoid organs where viral replication occurs, thereby allowing the flavivirus dissemination into the circulation and internal organs6.
ZIKV in pregnant women infects placental cells, causing their apoptosis and damage to vasculature causing placenta more permeable, facilitating entry into subsyncytial cytotrophoblasts17. Post-breaching of placental barrier, it replicates in various cell types including foetal macrophages and endothelial cells and infects the Hofbauer cells present in the villi of foetal capillaries. These infected cells can function as a reservoir of ZIKV replication and finally disseminate the virus into foetal blood18.
ZIKV has the ability to target human foetal brain causing cell death in various regions of the brain leading to microcephaly. The presence of ZIKV has been shown in cortical neural progenitors, radial glial cells and newly differentiating neural progenitors of developing mice1920. It also infects microglial and astroglial cells2122. In vitro studies suggest that ZIKV can infect human microglial cell line (CHME3), human primary astrocytes, human neural progenitor cells, human brain organoids, mouse astrocytes and neural stem cells (NSCs) efficiently1823242526. The capability of virus in infecting almost all the cell types of the brain results in severe consequences on the foetal brain.
Receptors involved in ZIKV entry
The virus entry into the host cells is facilitated by the interaction of ZIKV structural E protein with host receptors. Several cell surface receptors are implicated in flavivirus entry into the various host cells types27. Among the primary candidate receptors, best characterized are αvβ3 integrins, C-type lectin receptors, phosphatidylserine receptors TIM and TAM222829. Flaviviruses are known to produce a wide variety of structurally different virions, and perhaps this is the reason for their possible infectivity to various tissues, and hence the ability to infect a variety of cell types in its hosts15.
Various receptors are suggested to be involved in ZIKV entry including DC-SIGN, AXL, Tyro and TIM-1/4 entry/adhesion factors. In a study conducted by Hamel et al30, expression of candidate receptors like DC-SIGN or AXL showed enhanced viral infection in HEK293T cell line. Similarily, Tyro3-expressing HEK293T cells were also found to be highly permissive for ZIKV at 24 h post-infection (hpi), whereas expression of TIM-1 or TIM-4 showed marginal effect on ZIKV entry into HEK293T cells. There are contradictory studies regarding the role of AXL receptor in ZIKV entry into the brain cells. Meertens et al31 reported that the AXL receptor was responsible for virus entry into human microglia and astrocytes. The proposed mechanism suggests that the ZIKV upon interaction with AXL receptor ligand Gas6 activates its kinase activity, which results in downregulation of interferon (IFN) signalling and facilitates infection. This study also described that ZIKV infection in human glial cells could be inhibited by blocking the AXL receptor activity, for example, MYD1, a synthetic AXL decoy receptor and AXL kinase inhibitor R428 successfully inhibited the ZIKV infection. In another study by Hastings et al32, it was suggested that TAM receptors-Tyro3, Axl and Mertk were not required for ZIKV entry into the various organs of the mouse. The study involved multiple knockout mice, namely AXL−/−, Mertk−/−, AXL−/−Mertk−/− and AXL−/−Tyro3−/− and showed that all these mutants could be infected efficiently with ZIKV suggesting that TAM receptor including AXL might not be required for ZIKV entry and infection in rodents, also indicating that these receptors could have relevance only to human and non-human primates. Besides, induced pluripotent stem cells (iPSCs) are resistant to ZIKV infection despite expressing high levels of TYRO3, a TAM receptor33.
In another independent study to identify the role of AXL in virus entry, Chen et al34 showed that human astrocytoma cell line U-251MG expressed high levels of AXL receptors and was more susceptible to ZIKV, whereas T98G, glial cell line, known to express low levels of AXL, showed low susceptibility to ZIKV. The virus induces cytopathogenic effects and apoptosis in U-251MG cell line. AXL knockout U-251MG cell line was resistant to ZIKV infection. However, these experiments suggested that ZIKV entry was independent of the AXL receptor. The physiological function of AXL receptor activation is to block IFN signalling; hence, its ablation causes enhanced IFN signalling in AXL knockout cells, which is responsible for resistance to ZIKV. The authors described an explicit finding that the AXL signalling promoted ZIKV infection but was not necessary for its entry34. It is essential to identify the candidate receptors to decipher mechanisms of ZIKV neurotropism and its pathogenesis to discover new approaches toward designing therapeutics and vaccines against the virus.
Complications in ZIKV infection
Zika fever: The incubation period of flavivirus is around 2-7 days after the mosquito bite. In adults, one in five patients infected with ZIKV show symptoms35. In contrast with dengue and chikungunya fever, the onset of symptoms is difficult to assess in zika fever since there is no abrupt clinical onset3637. The mild intensity illness lasts from a span of days till weeks in case of adult infections. No haemorrhagic events associated with zika fever have been reported to date, distinctive in other flavivirus fever episodes.
Guillain–Barré syndrome: Guillain–Barré syndrome (GBS) is an acute self-limited polyneuropathy. GBS is a demyelinating disease of the peripheral nervous system, also referred to as acute inflammatory demyelinating polyneuropathy. It represents a range of symptoms from a very mild case with a brief weakness to devastating paralysis, leaving the person unable to breathe independently. The likely cause of the onset of GBS pertains to an autoimmune process38.
GBS has also been observed in other arboviral infections, such as Dengue virus (DENV) and Chikungunya virus (CHIKV)3940 but is a rare event. The causal factors rendering GBS and ZIKV infection need extensive explorations. ZIKV-induced GBS has been transient in duration, and most patients fully recover. No deaths were reported during the French Polynesian outbreak due to GBS41. Other possible neurological complications in ZIKV-infected adult individuals have been covered by Brito Ferreira et al42.
Congenital zika syndrome (CZS): In a study of pregnant women with ZIKV infection, abnormally high rates of early miscarriages and intrauterine foetal demise were reported. In about 20 per cent of ZIKV-infected pregnancies, impaired foetal growth and placental dysfunction have also been reported4344. Foetuses born with ZIKV infection have congenital zika syndrome (CZS).
CZS comprises a recognizable pattern of structural anomalies and functional disabilities in central and peripheral nervous system. By definition, severe microcephaly represents head circumference below the third percentile for sex and age45. High risk of severe microcephaly cases has been reported from 1 to 15 per cent in confirmed prenatal ZIKV infections4647. Manifestations of ZIKV-induced severe microcephaly are predominantly neurological. Imaging studies have also described cortical disorders, ventriculomegaly, lissencephaly, cerebral calcifications, periventricular, cortical and subcortical zones calcifications, cortical development errors, hydrocephaly and pachygyria/agyria4849. Craniofacial disproportion is also observed in the affected newborn50. Neurological anomalies are present in affected newborns later in life with normal head circumference also50.
In a clinical study, 141 children (age 1-14 yr) who suffered congenital ZIKV infection and in all the infants, structural brain abnormalities were detected by neuroimaging studies. These children were assessed for the prevalence of epilepsy. The incidence of epilepsy was high (67%), and the average age of infants at the onset of epilepsy was 4.9 months. In 74 per cent of infants, the seizures started to occur during the first six months of life. The main seizure types were epileptic spasms (72%), focal motor seizures (21%) and tonic seizures (4%)51. Another follow up study conducted by the National Center on Birth Defects and Developmental Disabilities (NCBDDD), Centers for Disease Control (CDC) described that 19 children were born with severe microcephaly, positive for ZIKV infection and most of them suffered from severe functional limitations such as motor impairment, seizure disorders, hearing and vision abnormalities and sleep difficulties (age group 19-24 months post-ZIKV infection)52.
Susceptibility of central nervous system (CNS) cells to ZIKV
Neural stem cells (NSCs): The ventricular zone (VZ), sub-VZ (SVZ) and hippocampal dentate gyrus are well-defined germinal niche of mitotically active NSCs. Given adequate cues, NSCs differentiate into neurons, astrocytes or oligodendrocytes and maintain NSC population by the process of self-renewal (symmetric cell division)53. Any effect on proliferation and differentiation properties of NSCs challenges neurodevelopmental processes. Numerous in vitro and in vivo studies have proved that NSCs are highly susceptible to ZIKV infection545556. The Table provides a summary of the effects of ZIKV on various brain cell types. Recent literature describes that induced pluripotent stem cells (undifferentiated form) are less permissive to ZIKV infections55; however, the cells differentiated from iPSCs such as NSCs are permissive to ZIKV30. Micrographs of infected neurospheres showed ZIKV bound to the plasma membrane, endoplasmic reticulum membranes, in mitochondria and vesicles of cells57. ZIKV induces apoptosis in NSCs and neurospheres. Growth areas of brain organoids from human iPSC also significantly reduced upon ZIKV exposure57. Fibroblast growth factor 2 (FGF2) signalling has been shown to be involved in replication of ZIKV in NSCs58. Li et al59 described that Asian strain of ZIKV (SZ01) infects embryonic mouse brain. The mouse embryo was infected at day 13.5 and inspected post three and five days of infection. ZIKV efficiently replicated in the mouse brain and targeted mainly SOX-2 (stem cell marker) and Ki67 (proliferation marker) positive cells. ZIKV infection induced proliferation arrest, apoptosis and impaired differentiation of NSCs leading to cortical thinning and hence resulting in microcephaly. The Global gene expression studies of ZIKV-infected brains revealed upregulation of candidate flavivirus entry receptors and dysregulation of genes associated with immune responses, cell death and microcephaly54.
| Cell type | Efficiency | Effects | Model system | Pathways/molecules involved | References |
|---|---|---|---|---|---|
| NSCs | +++ | Apoptosis, cell cycle arrest, inhibition of NSC differentiation, migration | In vivo mouse/human, in vitro mouse/human | CASP-3 activation, TLR3 signalling, autophagy, ER stress and UPR, dysregulation of mTOR signalling, mir-204-3p/PAX3 and mir-1273g-3p/NOTCH2 axis | 21556062727377 |
| Astrocytes | ++ | Activation, proliferation, apoptosis | In vivo mouse, in vitro human/mouse | UPR, autophagy | 232582 |
| Microglia | + | Activation | In vitro mouse, in vivo mouse | IFNα signalling, proinflammatory molecules - IL-6, MCP1, TNFα, IL1β, IL8, MIP-1α, iNOS | 2572 |
| Oligodendrocytes | + | Apoptosis | In vitro mouse | - | 86 |
| Brain microvascular endothelial cells | ++ | Activation, apoptosis | In vivo mouse in vitro human | IFNβ, IFNλ, interferon stimulated genes - IFIT1, ISG15, IFIT2, IL6, CCL5 | 878889 |
| Neurons-CNS | + | Apoptosis | In vivo human/mouse, in vitro human/mouse | CASP-3 activation | 5576 |
| Neurons-PNS | ++ | Apoptosis | In vivo mouse, in vitro mouse | CASP-3 activation | 778081 |
+, ++, +++ depicts the degree of susceptibility of cells towards ZIKV. CNS, central nervous system; PNS, peripheral nervous system; NSC, neural stem cell; TLR3, toll-like receptors-3; UPR, unfolded protein response; CASP-3, Caspase-3
ZIKV reduces expression of adherens junction proteins which help in anchoring radial glial cells (RGCs) and alter their properties in mouse brain. These adherens junction proteins include ZO-1 (zonula occludens-1), SMAD7 (β-catenin, SMAD family member 7) and NUMBL (NUMB like endocytic adaptor protein). ZIKV NS2A protein, when expressed in developing brain, also led to a similar reduction in adherent junction proteins60. ZIKV impairs brain development by inducing apoptosis in NES (neuroepithelial stem) cells and RGCs. Another study discovered induction of caspase-1 and gasdermin D (GSDMD)-mediated pryroptotic cell death in neural progenitor cells, in response to ZIKV infection61.
ZIKV induces mitosis defects in human NSCs including formation of multipolar spindle, chromosome laggards, smaller nuclei and death of the progeny following cell division. An increased frequency of aneuploidy, such as monosomy, trisomy and polyploidy was observed in infected cells6263. ZIKV infection perturbed centrosome (the hallmark of premature differentiation) culminating into depletion of stem cell niche, impaired VZ, disrupted neurogenesis and ultimately cortical thinning64. DNA damage response has also been observed in ZIKV infected NSCs65. Global transcriptome analysis (RNA-seq) of ZIKV infected human NSCs derived from iPSCs showed a drastic change in the transcriptome of virus-infected NSCs66. Gene ontology (GO) analysis of differentially expressed transcripts highlighted the involvement of downregulated transcripts in cell-cycle-related pathways and upregulated transcripts in transcription, protein transport and catabolic processes55. In an independent study, transcriptome analysis of human embryonic stem cells derived brain organoids showed activation of innate immune response post-ZIKV infection67. Toll-like receptors-3 (TLR3) were upregulated in ZIKV infected brain organoids, and the crucial role of TLR3 in ZIKV neuropathogenesis has been predicted. The growth of ZIKV infected neurospheres and organoids could be rescued with TLR3 competitive inhibitor (thiophenecarboxamide propionate), confirming the role of TLR367. Two major immune pathways, nuclear factor κB (NF-κB) and signal transducer of activators of transcription (STAT), are highlighted by transcriptomic analysis. Activation of these two pathways is responsible for the increase in inflammatory molecules post-host-virus interaction; the study points to chronic inflammatory conditions of the brain post-ZIKV infection66.
Autophagy plays a pro-viral role in ZIKV replication. The virus induces autophagy in human foetal NSCs as indicated by the microtubule-associated protein 1A/1B-light chain 3, LC3-I to LC3-II conversion62. Mouse embryonic fibroblasts (MEF) with autophagy-related gene 3 (Atg3), an essential component for autophagy, knockout showed reduced ZIKV replication as compared to wild-type MEFs suggesting that ZIKV exploits host autophagosome machinery for its replication68. ZIKV proteins NS4A and NS4B when co-overexpressed in human foetal NSCs, resulted in reduced neurosphere growth and induced autophagy in cells. NS4A and NS4B also impaired differentiation of NPCs into neurons and astrocytes in vitro68. ZIKV replication suppressed phosphorylation of Akt at Thr308 and Ser473, hence reducing mTOR phosphorylation at Ser2448. Similar response was found when NS4A and NS4B were expressed together68. Sofosbuvir, an Food and Drug Administration (FDA) approved antiviral drug, protects NES cells from ZIKV-induced cell death, perhaps because the compound also inhibits the re-localization of pTBK1 to mitochondria (pTBK1 plays a significant role in inducing innate antiviral immunity and cell proliferation)69. ZIKV infection also alters secreted proteome of mouse NSCs. Specific proteins crucial during repair or neuronal damage and proteins that antagonize and promote infection are found to be upregulated in the secretome of ZIKV infected NSCs. TLR3 was also upregulated in NSCs post-ZIKV infection in secretome70.
Overexpression of ZIKV E protein in human foetal NSCs (fNSCs) reduces proliferation and leads to cell-cycle arrest at the G0/G1 phase. E protein induced apoptosis in differentiating NSCs at day three and hampered the migration of newly differentiating cells from neurospheres71. Similar effects were observed in the mouse brain in the SVZ region when E protein was electroporated in utero at embryonic day 13.5 and analyzed at embryonic day 15. It was observed that proliferation was attenuated as assessed by a significant decrease in Ki6771.
MicroRNA (miRNA) are non-coding regulatory small RNA (19-24 nucleotide long) molecules. These molecules downregulate their target genes by binding to the 3’ UTR of the target transcripts. At the molecular level, E protein altered global miRNA profile of human foetal NSCs71. At least 25 miRNAs were differentially expressed on E protein overexpression71. mir-1273g-3p and mir-204-3p were highly expressed in response to E protein in NSCs, interestingly, mir-1273g-3p and mir-204-3p are molecular targets of important development-related genes paired box gene 3 (PAX3) and notch receptor 2 (NOTCH2), respectively71. This study provided compelling data to suggest the crucial role of ZIKV E protein and miRNA circuitry in ZIKV neuropathology.
In human infections, ZIKV was found to trigger endoplasmic reticulum (ER) stress and unfolded protein response (UPR) in the cerebral cortex of ZIKV-infected post-mortem foetuses72. A similar response was also observed in cultured human NSCs and mouse embryos72. ER stress markers – calnexin, calreticulin, MX, PDI1, PERK–ATF4 and ratio of spliced/unspliced XBP1 were upregulated post-ZIKV infection in fNSCs and mouse brain72. Induction of DNA methylation was observed in human cerebral organoid-derived neural precursor cells, astrocytes and neurons following ZIKV infection73. ZIKV was also found to modulate long non-coding profile of human NSCs and alternate splicing pattern74.
ZIKV modulates the fate of NSCs in the developing brain. ZIKV infection increased glial fibrillary acidic protein (GFAP) and aldehyde dehydrogenase 1 family member A (ALDH1a)- markers of astrocytic differentiation, in mouse NSCs indicating differentiation of NSCs towards glial lineage70. Induction of glial differentiation could be to initiate an immune response against ZIKV70. ZIKV proteins NS4A and NS4B when co-overexpressed in NSCs also inhibited neural differentiation68. Moreover, ZIKV E protein also induced massive apoptosis in differentiating human foetal NSCs into neurons71. ZIKV NS5 is localized to the nucleus and during mitosis, re-localized to centrosomes. Cells expressing NS5 took longer to complete the cell cycle, suggesting that ZIKV induced cell cycle arrest by translocation of NS5 to the nucleus and by interacting with several centrosome proteins75. Hence, the virus has the capability to deregulate the fate of NSCs that may impact the overall pool of brain cells.
Neurons: Lack of functional neuron and impaired neurogenesis are hallmarks of ZIKV neuropathogenesis. The primary target of ZIKV is NSCs; however, ZIKV can infect mature and immature neurons in vitro as well as in vivo in mouse and human model systems though at low efficiency557677. ZIKV targets newly forming special AT-rich sequence-binding protein 2 (SATB2+) neurons78. Human foetal brain tissues ageing from 14 to 21 gestation weeks with ZIKV infection (Strain MR766) were found to have many neurons affluent areas positive for ZIKV. The virus infects Tbr2-expressing intermediate progenitor cells and microtubule-associated protein 2 (MAP2+) mature neurons to some extent79.
ZIKV, strains MR766 and IbH30656 were found to infect sensory neurons of the trigeminal and dorsal root ganglia but without inducing cell death. Primary adult murine sympathetic neurons of the superior cervical ganglia and parasympathetic neurons of the ciliary ganglia did not show signs of active ZIKV76. Human iPSCs derived cortical neurons as well as the motor neurons are susceptible to ZIKV infection (by Strains: MR766 from the African lineage and PRVABC59 from Asian lineage), where Asian lineage showed an almost ten-fold replication rate80. Another study explored ZIKV infection in the peripheral nervous system (PNS), both in vivo and in vitro model systems. Peripheral neurons were permissive to the infection leading to cell death77.
Astrocytes: Astrocytes are associated with a plethora of roles including formation of blood-brain barrier (BBB) and its maintenance, synaptogenesis, repair of tissue damage, neurotransmission, metabolic regulation, innate and adaptive responses moderation to pathogens and supporting the synaptic transmission. Astrocytes are thought to be an early target of ZIKV during in utero infections due to their participation in BBB formation. ZIKV infects astrocytes with high propensity23268182. Astrocytes respond to all forms of central nervous system (CNS) damage and disease resulting in changes in their gene expression, cellular structure and function. Such responses of astrocytes are referred to as astrogliosis. Astrogliosis is identified as a major contributor to CNS disorders, including viral infections. During ZIKV infection, astrogliosis and the associated inflammatory scenario may lead to massive cell death in the developing brain. Astrogliosis has been observed in ZIKV-infected human foetuses25. GFAP is a hallmark of glial activation, robust upregulation of GFAP is observed in ZIKV infected mouse brain25. ZIKV also inhibits activation of NLRP3-dependent inflammasome in bone marrow-derived macrophages (MDMs) and mixed glial cells from mouse brain83. The susceptibility of astrocytes could be due to high expression of AXL, TAM family receptor, but more pieces of evidence are needed to claim AXL as candidate entry receptor for ZIKV. Furthermore, satellite glial cells which surround and protect the soma of ganglionic neurons in peripheral sensory and autonomic ganglia can be infected with ZIKV; the virus was found to kill satellite glial cells in vitro76. ZIKV susceptibility of astrocytes results in progressive cell death inducing necroptosis in infected cells. It is indicated by increased phosphorylation of receptor-interacting serine/threonine- protein kinase (RIPK)1, RIPK3, and mixed lineage kinase domain-like (MLKL) protein. It was found that ZIKV-induced necroptosis was RIPK3-dependent. Human astrocytes when pre-treated with an inhibitor for RIPK3 activation, showed restricted viral replication. This study suggested that induced necroptosis could play a protective role in viral pathogenesis84.
As in NSCs, ZIKV modulates miRNA circuitry in astrocytes. Kozak et al23 described the differential expression of miRNA post-ZIKV infection in SVG human astrocyte cell line. Several miRNAs were differentially expressed at 24 h post-infection; 11 miRNAs were found to be upregulated by more than 2-fold. At 48 h post-infection, three miRNAs - miR-411-3p, miR-323a-5p and miR-194-5p were upregulated by more than 2-fold, and at 72 h post-infection, miR-9-5p was upregulated over 7-fold. These observations highlighted that miRNAs might have a role to play in ZIKV neuropathology. GO analysis of targets of differentially expressed miRNAs in SVG cell line (human astrocyte) are involved in viral life cycle, viral transcription, cell cycle, immune system response and innate immune response. Fig. 3 is a schematic representation of the role of miRNAs in ZIKV-induced alterations in NSCs and astrocytes.
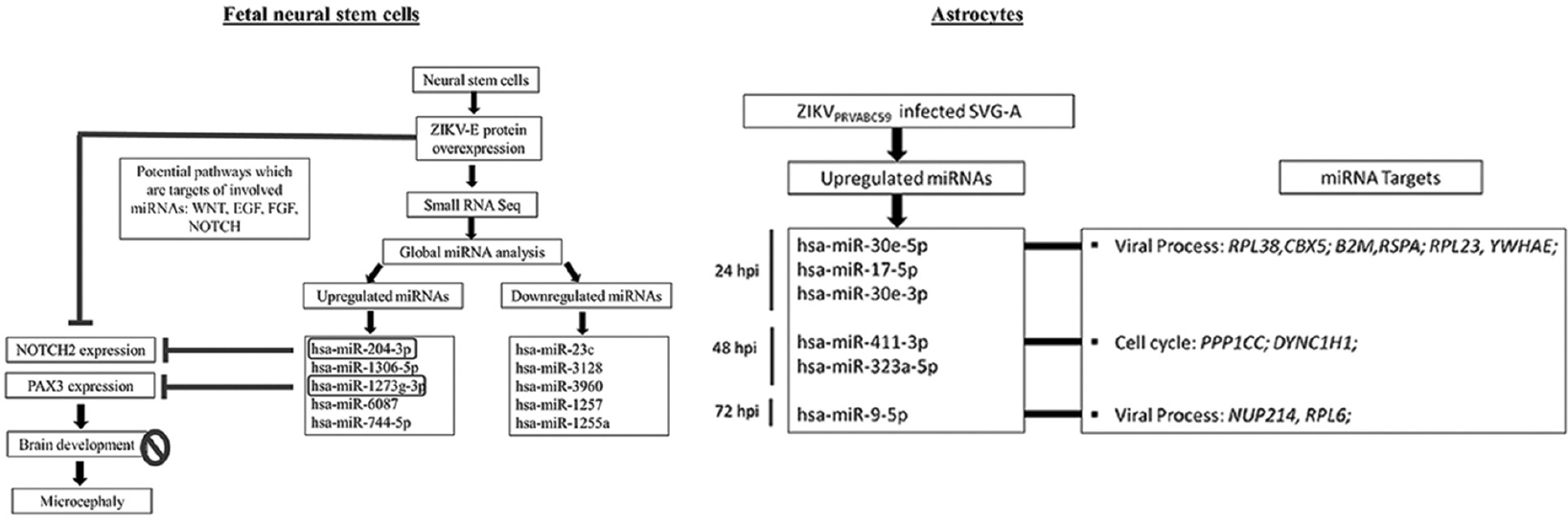
- Role of microRNAs in the zika virus-induced alterations in neural stem cells and astrocytes. In human foetal neural stem cells, zika virus E protein disrupted miRNA circuitry, as investigated through small RNA sequencing. NOTCH2 and PAX3, two significant development-related transcripts, were downregulated by miR-204-3p and miR-1273-3p 62, respectively. Altered expression of NOTCH2 and PAX3 may cause hindered brain development leading to ZIKV-associated microcephaly. Similarly, SVG-astrocytes infected with ZIKV (strain PRVABC59) resulted in altered miRNA circuitry at 24 hpi, 48 hpi and 72 hpi. Upregulated miRNAs were found to target genes involved in viral process and cell cycle.
Oligodendrocytes: Infection of oligodendrocytes is not pursued much till date; hence, minimal information is available regarding the role of oligodendrocytes during ZIKV infection. Since oligodendrocytes are essential for the functioning of neurons, alterations in their properties would impair neural functioning. Cumberworth et al85 reported that oligodendrocyte precursor cells and oligodendrocytes were susceptible to ZIKV infection inducing cell death in in vitro mouse cultures. ZIKV infected peripheral nervous system (PNS) cultures of neurons and Schwann cells with very low efficiency. Li et al25 also described that ZIKV caused loss of MBP-positive cells (a marker for oligodendrocytes) and CNP-positive cells (immature oligodendrocyte) in newborn mouse cortex.
Brain microvascular endothelial cells (BMECs): BMECs form the BBB providing a tight seal which imparts selective permeability across the BBB. BMECs, when experience insult, release cytokine and chemokines, which ultimately lead to the infiltration of monocytes into the brain. BMECs infected by ZIKV upon activation release inflammatory molecules –interferon (IFN)-β, IFN-λ, interferon induced protein with tetratricopeptide repeats (IFIT)1, IFIT2, interferon-stimulated gene (ISG)15, interleukin (IL)-1 and IL-68687. The viability of BMECs remains unaffected in in vitro model systems of BBB8688. NS5 sumoylation has been found to be associated with persistent ZIKV infection in human BMECs89. Though effects of ZIKV on BMECs remain mild, the virus creates an inflammatory scenario90.
Microglia: Microglial cells play a pivotal role during the brain development and significantly contribute in neuroinflammation and neuropathogenesis in viral infections. It plays additional roles in synapse formation, brain inflammatory response, neurodegenerative diseases, stroke, trauma and regeneration. ZIKV has been shown to target microglia, primary human monocytes and MDMs thereby altering their properties229192. ZIKV induced an anti-viral response by enhancing IFN-α expression and other pro-inflammatory molecules, i.e., IL-1β, IL-6, IL-8, macrophage inflammatory protein (MIP) -1α, MIP-1β, monocyte chemoattractant protein (MCP)-1, tumour necrosis factor alpha (TNF-α), IL-1Rα, IL-1α and IL-12p702291.
Pro-inflammatory molecules released from astrocytes impair the properties of NSCs and neurons. In vitro studies suggested that conditioned medium from ZIKV-infected microglia hampered the growth of neurospheres and proliferation of NSCs. Moreover, conditioned medium from infected microglial cells reduced neurogenesis and enhanced astrogliogenesis. TNF-α and IL-6 cytokines were preeminent among others, as blocking them in conditioned medium rescued neurogenesis and inhibited enhanced astrogliogenesis92. Furthermore, the role of microglia is evident as infected microglial cells appear to be phagocytosing pyknotic nuclei post-ZIKV infection, suggesting chronic inflammatory conditions in ZIKV infected brains85. Investigations into the detailed molecular pathways induced by ZIKV in microglial cells are hence necessary.
Open questions
In spite of the recent advances in the understanding of mechanisms that may underlie ZIKV neuropathogenesis, basic and clinical researchers are striving to find answers for some open questions in a hope to reduce the burden of consequences arising following ZIKV infections. Some of the open questions are as follows - What makes ZIKV infect such a wide variety of cell types? Which are the receptors/co-receptors and what are the mechanisms involved in the entry of virus into the host cells? What are the receptor-independent (if any) mechanisms of ZIKV invasion? How did the virus gain neurotropism? What all signalling pathways are involved in disrupting the proliferative, differentiating and migrating properties of NSCs? What can be the possible regimen for blocking/treating intrauterine ZIKV infection?
Remarks & conclusions
ZIKV being an arbovirus with the existence of various lineages, can continue to stay a global threat, especially in densely populated developing countries that lack health awareness. The virus can infect multiple cell types of brain as well as other organs of the body. Sustained efforts for prevention and clinical management of ZIKV related diseases are critical.
The virus modulates host cell machinery for its successful replication. The virus and its proteins inhibit innate antiviral response by modulating TLR3 activity; it also enhances autophagosome formation in cells for its replication. Once the virus enters brain, it deteriorates development of the brain at various levels: (i) induction of apoptosis and hampered proliferation in early progenitors, (ii) inhibition of neurogenesis and astrogliogenesis, (iii) activation of brain immune cells (astrocytes and microglia) to release many cytokines and chemokines. The virus causes massive cell death in the developing brain. The damage caused to the brain in utero is irreversible and leaves the newborn with various severe motor and cognitive disabilities.
Financial support & sponsorship: The first author (RB) acknowledges the Council of Scientific & Industrial Research, New Delhi for providing junior and senior research fellowships. The authors also acknowledge the support of the facilities provided under the Biotechnology Information System Network (BTISNET) grant, Department of Biotechnology (DBT), India, and Distributed Information Centre at NBRC, Manesar, India.
Conflicts of Interest: None.




