
Translate this page into:
Long-range PCR amplification-based targeted enrichment & next generation sequencing: A cost-effective testing strategy for lysosomal storage disorders
For correspondence: Dr Prajnya Ranganath, Department of Medical Genetics, Specialty Block, Fourth Floor, Nizam’s Institute of Medical Sciences, Panjagutta, Hyderabad 500 082, Telangana, India e-mail: prajnyaranganath@gmail.com
-
Received: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
Lysosomal storage disorders (LSDs) are genetic metabolic disorders which result from deficiency of lysosomal enzymes or defects in other lysosomal components. Molecular genetic testing of LSDs is required for diagnostic confirmation when lysosomal enzyme assays are not available or not feasible to perform, and for the identification of the disease causing genetic variants. The aim of this study was to develop a cost-effective, readily customizable and scalable molecular genetic testing strategy for LSDs.
Methods:
A testing method was designed based on the in-house creation of selective amplicons through long range PCR amplification for targeted capture and enrichment of different LSD genes of interest, followed by next generation sequencing of pooled samples.
Results:
In the first phase of the study, standardization and validation of the study protocol were done using 28 samples of affected probands and/or carrier parents (group A) with previously identified variants in seven genes, and in the second phase of the study, 30 samples of enzymatically confirmed or biopsy-proven patients with LSDs and/or their carrier parents who had not undergone any prior mutation analysis (group B) were tested and the sequence variants identified in them through the study method were validated by targeted Sanger sequencing.
Interpretation & conclusions:
This testing approach was found to be reliable, easily customizable and cost-effective for the molecular genetic evaluation of LSDs. The same strategy may be applicable, especially in resource poor settings, for developing cost-effective multigene panel tests for other conditions with genetic heterogeneity.
Keywords
Long-range polymerase chain reaction-based targeted amplification
lysosomal storage disorders
molecular genetic testing

Lysosomal storage disorders (LSDs) are a group of over 50 inborn errors of metabolism characterized by intra-lysosomal accumulation of complex macromolecules, which result from the deficiency of lysosomal enzymes or from defects in key lysosomal membrane proteins, proteins involved in lysosomal enzyme trafficking or lysosomal enzyme activator proteins1,2. With a cumulative prevalence of around one in 5000 live births, they pose a sizeable health burden worldwide and are associated with significant morbidity and mortality related to neurological, visceral, cardiovascular and skeletal involvement1,3.
Accurate diagnosis is essential for the proband as well as for the family, for appropriate management, prognostication, surveillance, genetic counselling and prenatal/pre-implantation genetic testing. Conventionally, LSDs have been diagnosed through lysosomal enzyme assays and these assays continue to be the gold standard diagnostic test for affected probands. However, in addition to issues inherent to biochemical diagnosis such as erroneous results due to sample degradation resulting from improper handling and lack of temperature control during transportation, enzyme assays have other limitations with respect to the diagnosis of LSDs4. Enzyme assays are not reliable for carrier testing as the enzyme levels in carrier individuals overlap with the normal range. Prenatal genetic testing based on enzyme assay may sometimes give equivocal results, especially in carrier fetuses or when there is maternal cell contamination5. Due to phenotypic variability and overlap in the clinical manifestations of different LSDs, multiple enzyme assays may be required in some cases, to establish the diagnosis, which can be time-consuming, laborious and expensive and would also require a larger volume of blood for testing2. For LSDs which are not caused by deficiency of enzymes but by transporter defects [e.g. Niemann-Pick disease (NPD) type C] or activator protein deficiencies (e.g. sphingolipid activator protein deficiency), confirmation of the diagnosis requires molecular testing of the gene encoding the specific protein2. A common panel providing mutation analysis for multiple LSDs in one single reaction would thus serve as a cost-effective and time saving diagnostic tool for such clinically indistinguishable LSDs, LSDs with overlapping phenotypes and LSDs not diagnosable through enzyme assays6,7.
The Sanger sequencing technique is time consuming and expensive for multigene panel testing. Next generation sequencing (NGS) technologies have high throughput and make it possible to sequence multiple genes and large genomic regions by performing several millions of accurate sequence reactions in parallel. For sequencing targeted regions of the genome through NGS, target enrichment has to be done through capture kits that enable ‘capture’ of the desired genomic regions8,9. The commercially available target enrichment kits which work chiefly on the principle of solution-based capture or array-based capture are expensive and increase the cost of the test and they are not easily customizable and scalable for small groups of genes, for example, a group of genes associated with a specific phenotype such as dysostosis multiplex-associated LSDs.
The objective of this study was to design and validate a strategy of long range PCR (LR-PCR) based targeted enrichment, followed by NGS, for molecular genetic testing of LSDs and to evaluate the cost-effectiveness, scalability and customizability of this technique.
Material & Methods
The study was conducted over three years from November 2014 to November 2017 at the Diagnostics Division of the Centre for DNA Fingerprinting and Diagnostics (CDFD), Hyderabad after approval of the Institutional Ethics Committee. Individuals across all age groups who were diagnosed to have an LSD based on the clinical features and imaging findings and in whom the specific diagnosis was confirmed by either enzyme assay and/or molecular genetic testing and/or their unaffected carrier parents, were included in the study, after obtaining a written informed consent.
Peripheral blood samples were collected for all individuals recruited into the study and DNA was extracted from the blood samples as described below. The study samples were classified into group A and group B. Group A consisted of affected individuals and/or parents who had already undergone molecular genetic testing (with or without enzymatic confirmation) through Sanger sequencing, and in whom, the disease causing gene variants were already identified. The DNA samples of this group were used for the first phase of the study, which involved standardization and validation of the study technique. Group B consisted of patients who had an enzymatically confirmed or biopsy-proven diagnosis of a specific LSD and/or their unaffected carrier parents, in whom molecular genetic testing had not been done already. The DNA samples of this group were used in the second phase of the study. The samples of probands, as well as carrier parents, were collected, to validate the utility of the method for detection of homozygous as well as heterozygous variants.
Sample collection and DNA extraction: Three millilitres of peripheral blood was collected in an EDTA vacutainer tube, from each study participant. Genomic DNA was extracted from each blood sample using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) and the DNA was quantified using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Massachusetts, USA).
Long range PCR (LR-PCR): The primers for LR-PCR were designed using the Primer3 software version 4.0.0 (http://bioinfo.ut.ee/primer3) for the lysosomal storage disorder-associated genes, as listed in Table I. A total of 22 genes were included (NEU1, ARSA, SMPD1, IDUA, NPC1, NPC2, GNPTAB, GNPTG, GAA, GALNS, GLB1, MCOLN1, GBA, NAGLU, SGSH, IDS, HEXA, HEXB, GLA, GALC, ASAH1 and GUSB). Each set of LR-PCR primers was designed to amplify around 5-10 kb fragments. For genes smaller than 10 to 15 kb in size (including the complete exonic and intronic regions), the entire gene could be covered within a single LR-PCR fragment, whereas for genes larger than 15 kb in size, from two to up to nine sets of LR-PCR primers had to be designed to cover the entire gene, depending on the size of the gene, as mentioned in the Supplementary Table. The LR-PCR primer sets were designed to completely cover the exonic regions, along with the flanking intronic sequences (up to at least 500 bases on both sides of each exon), and at least 500 bases upstream to the 5´ end and 500 bases downstream to the 3´ end of each of the 22 genes. In addition, all the introns that got included as intervening sequences, within the proximal and distal exonic extent of each LR-PCR fragment, were also completely covered, as detailed in Supplementary Table. LR-PCR primers were not designed for exons 1 and 2 of GNPTAB, exon 1 of HEXA, exon 1 of HEXB, exon 1 of NPC2 and exon 1 of ASAH1, as these exons were small and could be covered through short PCR and Sanger sequencing, and their flanking intronic sequences were too large to enable them to be clubbed together with the next exon within one LR-PCR fragment. LR-PCR was standardized and performed using PrimeSTAR GXL DNA polymerase (Takara Bio Inc., Shiga, Japan). Agarose gel electrophoresis with ethidium bromide staining of the LR-PCR products was performed to demonstrate specific fragments ranging in size from 5-10 kb for the different LSD-associated genes included in the study.
| Sample ID | Affected status | Library number | Name of the gene | LR-PCR fragment in which variant (s) identified | Sequence variant previously identified through Sanger sequencing (DNA notation) | Sequence variant previously identified through Sanger sequencing (protein notation) | Zygosity | Position of the variant | Known/Novel Variant | Variant classification |
|---|---|---|---|---|---|---|---|---|---|---|
| A_001 | Affected proband | Lib_1 | NEU1 | NEU1-1 (Exons 1 to 6) | c.679G>A/ c.200delG | p.Gly227Arg/ p.Ser67ThrfsTer71 | CH | Exonic/Exonic | Known (reported in HGMD and ClinVar)/Novel | LP/LP |
| A_002 | Affected proband | Lib_1 | IDUA | IDUA-2 (Exons 3 to 14) | c.767T>G | p.Leu256Arg | Hom | Exonic | Novel | LP |
| A_003 | Affected proband | Lib_1 | ARSA | ARSA-1 (Exons 1 to 8) | c.327+1C>T/ c.338T>C | NA/p.Leu113Pro | CH | Splice site/Exonic | Novel/Novel | LP/VOUS |
| A_004 | Affected proband | Lib_1 | GAA | GAA-3 (Exons 16 to 20) | c.2783A>G | p.Tyr928Cys | Hom | Exonic | Novel | VOUS |
| A_005 | Affected proband | Lib_1 | GNPTAB | GNPTAB-3 (Exons 8 to 11) | c.1144A>C | p.Tyr382Pro | Hom | Exonic | Known (reported in ClinVar) | LP |
| A_006 | Unaffected carrier parent | Lib_1 | NPC1 | NPC1-8 (Exons 15 to 20) | c.2738delG | p.Gly913Ala fsTer23 | Het | Exonic | Novel | LP |
| A_007 | Affected proband | Lib_1 | GLB1 | GLB-1 (Exon-1) | c.75+2dupT | NA | Hom | Splice site | Known (reported in ClinVar) | LP |
| A_008 | Affected proband | Lib_2 | NEU1 | NEU1-1 (Exons 1 to 6) | c.880C>T/ c.194G>A | p.Arg294Cys/p.Trp65Ter | CH | Exonic/Exonic | Known (reported in HGMD)/Novel | LP/P |
| A_009 | Affected proband | Lib_2 | IDUA | IDUA-1 (Exons 1 to 2) | c.779C>T | p.Ser260Phe | Hom | Exonic | Novel | VOUS |
| A_010 | Affected proband | Lib_2 | ARSA | ARSA-1 (Exons 1 to 8) | c.901C>T/c. 417C>T | p.Arg301Trp/p.Pro139Pro | CH | Exonic/Exonic (splice site change) | Known (reported in HGMD)/Novel | LP/LB |
| A_011 | Unaffected carrier parent | Lib_2 | GAA | GAA-3 (Exons 16 to 20) | c.2783A>G | p.Tyr928Cys | Het | Exonic | Novel | VOUS |
| A_012 | Affected proband | Lib_2 | GNPTAB | GNPTAB-3 (Exons 3 to 5) | c.440delC | p.Asn148ThrfsTer4 | Hom | Exonic | Novel | P |
| A_013 | Unaffected carrier parent | Lib_2 | NPC1 | NPC1-7 (Exons 12 to 14) | c.2068A>T | p.Ile690Phe | Het | Exonic | Novel | VOUS |
| A_014 | Unaffected carrier parent | Lib_2 | GLB1 | GLB-1 (Exon-1) | c.68G>C | p.Gly23Ala | Het | Exonic | Novel | VOUS |
| A_015 | Affected proband | Lib_3 | NEU1 | NEU1-1 (Exons 1 to 6) | c.679G>A | p.Gly227Arg | Hom | Exonic | Known (reported in HGMD and ClinVar) | LP |
| A_016 | Affected proband | Lib_3 | IDUA | IDUA-2 (Exons 3 to 14) | c.972+1G>A | NA | Hom | Splice site | Known (reported in ClinVar) | P |
| A_017 | Affected proband | Lib_3 | ARSA | ARSA-1 (Exons 1 to 8) | c.737G>A | p.Arg246His | Hom | Exonic | Known (reported in ClinVar) | LP |
| A_018 | Unaffected carrier parent | Lib_3 | GAA | GAA-3 (Exons 16 to 20) | c.2783A>G | p.Tyr928Cys | Het | Exonic | Novel | VOUS |
| A_019 | Affected proband | Lib_3 | GNPTAB | GNPTAB-3 (Exons 3 to 5) | c.571G>A | p.Val191Ile | Hom | Exonic | Known (reported in ClinVar) | LP |
| A_020 | Unaffected carrier parent | Lib_3 | NPC1 | NPC1-8 (Exons 15 to 20) | c.2378delA | p.Gly913Ala fsTer23 | Het | Exonic | Novel | LP |
| A_021 | Affected proband | Lib_3 | GLB1 | GLB1-6 (Exons 13 to 15) | c.1445G>A | p.Arg482His | Hom | Exonic | Known (reported in ClinVar and HGMD) | P |
| A_022 | Affected proband | Lib_4 | NEU1 | NEU1-1 (Exons 1 to 6) | c.887A>G | p.Tyr296Cys | Hom | Exonic | Novel | VOUS |
| A_023 | Affected proband | Lib_4 | IDUA | IDUA-2 (Exons 3 to 14) | c.895G>T | p.Glu299Ter | Hom | Exonic | Novel | P |
| A_024 | Unaffected carrier parent | Lib_4 | SMPD1 | SMPD1-1 (Exons 1 to 6) | c.1492C>T | p.Arg498Cys | Het | Exonic | Known (reported in ClinVar) | LP |
| A_025 | Affected proband | Lib_4 | GAA | GAA-1 (Exons 1 to 2) GAA-3 (Exons 15 to19) | c.189delA/c. 2122delC | p.Arg66Gly fsTer76/p.His708Thr fsTer56 | CH | Exonic/Exonic | Novel/Novel | P/P |
| A_026 | Unaffected carrier parent | Lib_4 | GNPTAB | GNPTAB-3 (Exons 3 to 5) | c.440delC | p.Asn148ThrfsTer4 | Het | Exonic | Novel | LP |
| A_027 | Unaffected carrier parent | Lib_4 | NPC1 | NPC1-8 (Exons 15 to 20) | c.2604+1G>A | NA | Het | Splice site | Known (reported in ClinVar) | P |
| A_028 | Affected proband | Lib_4 | GLB1 | GLB1-2 (Exons 2 to 5) GLB1-6 (Exons 13 to15) | c.522T>G/c. 1242delG | p.Tyr174Ter/p.Phe415Leu fsTer46 | CH | Exonic/Exonic | Novel/Novel | P/P |
Hom, homozygous; Het, heterozygous; CH, compound heterozygous; P, pathogenic; LP, likely pathogenic; VOUS, variant of uncertain significance; LB, likely benign; HGMD, human gene mutation database; PCR, polymerase chain reaction; LR-PCR, long-range PCR
| Gene symbol and transcript id | Name of the disease | Size of the gene (bp) | LR-PCR primer sets | Exons covered by each primer set | PCR product size (bp) | Forward primer sequence | Reverse primer sequence |
|---|---|---|---|---|---|---|---|
| GBA ENST00000327247.9 | Gaucher disease | 11,440 | GBA-1 | 1 to 11 | 7765 | CGACTTTACA AACCTCCCTG | CCAGATCCTAT CTGTGCTGG |
| SMPD1 ENST00000342245 | Niemann-Pick disease A and B | 6967 | SMPD1-1 | 1 to 6 | 5996 | ACTACCCACTTCC CAGACGAGTTCA | TAGGAGCTGGGGA GGGAGAGATCTA |
| NPC1 ENST00000269228 | Niemann-Pick disease, Type C | 83,114 | NPC1-1 | 1 | 4971 | GACTTTCTCCTGCC CTCCTGTCTCCA | AGTCCCAAGTCAA GTGTCCTGGCCAC |
| NPC1-2 | 2 to 3 | 4227 | CACTGACCCCTCC CCTCCGCTGAATTT | GTGTGTCCTGGCCT TCAAGAGTCTCTG | |||
| NPC1-3 | 4 | 4362 | GACTCTCATCATGCA GTCCTTCCTCTCCCC | AGCTCCCCAATTACC CCTCCCTGACAGTAG | |||
| NPC1-4 | 5 to 6 | 4129 | CGTGCCCGGCCAA CAGTATTAGGTT | GCTGGGGATGGGA GGTGTATGTAAG | |||
| NPC1-5 | 7 to 9 | 5796 | CTTACATACACCT CCCATCCCCAGC | GGAGGGAGAGAGA ACAGTGAGAGGG | |||
| NPC1-6 | 10 to 11 | 4939 | CATTCCCTCTCACT GTTCTCTCTCCCTCCC | CAACCTGGCCTCCTA AGTCCTCTCTTCCCA | |||
| NPC1-7 | 12 to 14 | 4282 | CGTCACCACCAGCA GTCCATGAAACTTCC | ACTCACTCCTACTGTC CCAAGGTCACTCC | |||
| NPC1-8 | 15 to 20 | 4611 | GGGACAGTAGGAGTG AGTAGGGATTGTGGC | AACCCCGAGCAAAA TGACCACCTCTGAGC | |||
| NPC1-9 | 21 to 25 | 6167 | GCAGCCAGTTACCC ACGGAAGCCAGATAT | GGCCTTTACAGAGTG TCAGTGAGCGGATC | |||
| NPC2 ENST00000555619 | Niemann-Pick disease, Type C | 19,185 | NPC2-1 (Sanger) | 1 | 857 | TATTGGGTAG TGCGGGAGGA | GCAAAAGGAG TTGGAAGGGG |
| NPC2-2 | 2 to 5 | 7607 | GCCTGTAATCCTAG CACTTTGGGAGGCC | CTGTAATCCCTAAC CCCTACCTGCCCTC | |||
| ARSA ENST00000216124 | Metachromatic Leukodystrophy | 7819 | ARSA-1 | 1 to 8 | 7319 | CACGCACACAAC AGACACACCCTAA | TACCCAAGTGAGA GGAAGTGGAGCA |
| IDUA ENST00000247933 | MPS Type I | 19,909 | IDUA1 | 1 to 2 | 4673 | CACCTCAATTTCG TGGGTAGCTGGG | CATGTGTAGGAAG CAGCAGGAAGGG |
| IDUA2 | 3 to 14 | 5660 | CTCACTCCCTGT CGTATCCCCTTCA | GAAGAGACTGCGG CCTTGGTTCCTG | |||
| IDS ENST00000340855.10 | MPS type II (Hunter syndrome) | 45,307 | IDS-1 | 1 to 4 | 5719 | CAGTACAGTGTAGG GCTAGGATTCCATCTC | TACCTTCCACCTCAC TTCCAAATCCTCACC |
| IDS-2 | 5 to 7 | 11,105 | CATGCCGTGTATC TGTCTCTGACCTA | GTTCTGGCCCTGA ATTACATGCTCTG | |||
| IDS-3 | 8 to 9 | 10,828 | CATGAGTCAAAAGAC AGGTAGGCACAGGAC | CCTCTTTCTTTCTGG GGAGTCTCTGTACTG | |||
| SGSH ENST00000326317.10 | Mucopolysaccaridosis type IIIA (Sanfilippo disease A) | 15,408 | SGSH | 1 to 8 | 14,661 | GTGTCAGGAGAGG TCACTATGGGTCT | TGGCAAGCTCTAT TCCCTCATCTCCT |
| NAGLU ENST00000225927.6 | MPS type IIIB (Sanfilippo disease B) | 9478 | NAGLU | 1 to 6 | 8860 | TCCTCGAACCTCCTA GCCTGTTAGTTACTC | CAGTGACCTTCTCAT TTTGACAGACCCAGG |
| GALNS ENST00000268695.9 | MPS type IVA (Morquio A syndrome) | GALNS-1 | 1 | 5007 | CCCAGGGAAGAAG TAGAAAGAAAGCGG | GACCTTCAGCAAAC TCCAACAGACCTG | |
| GALNS-2 | 2 to 4 | 5355 | CTTCTAGAGCAA AGTCCTGGCCCAC | CTAGAGCACCCC GACATCCCTGAAC | |||
| GALNS-3 | 5 to 9 | 7311 | GTCGTCACTCTAA TCCCTGCCTCTG | CCACACAGTCATT ACCAGCACCCAC | |||
| 43,237 | GALNS-4 | 10 to 11 | 6472 | CAATTACTAGGGAGC AGAGGTGGGAGCAG | CTAAGACAGGGAGGA GCAGGACCAACATG | ||
| GALNS-5 | 12 to 14 | 9834 | GTCTTAGGGTTTCTG TAGGGATTCTGAGCG | CTTCCCCTTCCTTGT TCCTGATTCTGTCTC | |||
| GLB1 ENST00000307363.9 | GM1-gangliosidosis and MPS type IVB (Morquio B) | 101,820 | GLB1-1 | 1 | 4158 | GTAGAGACGGGGTTT CATCATACTGGTCAG | GTGAATGTCTGAGAG GAGCACGATCTTGAG |
| GLB1-2 | 2 to 5 | 8330 | GATGAAGAGAGGTA GGGTTAGAGATGGGAC | GAAGAAGGAGAATGG GGCAGGAAGTAAGAG | |||
| GLB1-3 | 6 to 9 | 8098 | GGGGAGTGTGTGGG TCTGTGTAAATCTAGA | GAGGGAGAGTGTGG AATGAATGCTAATGGG | |||
| GLB1-4 | 10 | 5319 | CTAGAGGAGAGCAGG GAGAGAAAGGCATC | GACTAGAGAGGGAC ATTAGAGGGGCTTCC | |||
| GLB1-5 | 11 to 12 | 5543 | GAGGATTGCATACA TCACGGCCTTTACC | GGAAAGCCTCAA ACAATCAGCCTCAG | |||
| GLB1-6 | 13 to 15 | 7241 | CTCAAGAGATCCTCC CCAAGTAACTGAGAC | CCTGTTCGTATTCCT TACCCTGGTCTTCTC | |||
| GLB1-7 | 16 | 5558 | CAAAGACTTTCCC TTCCTAGAGCCTGTG | CGGCAGGATGACA GTATATTCCTCAGTG | |||
| GUSB ENST00000304895.8 | MPS type VII (Sly disease) | 22,830 | GUSB-1 | 1 to 4 | 5820 | TGATGTGTAGGG ATTCACCACCC | TGCTCTATGGTG CATTGTCTTTGC |
| GUSB-2 | 5 to 8 | 4098 | ACACAGAATT CAGGACAGGC | GTGTGGTGGT TCACACCTGT | |||
| GUSB-3 | 9 to 11 | 8450 | GAGCAGGTGTTTG AGGCTTCTTTGG | GGATTAAACCAG CTTCCCCAACTTT | |||
| GUSB-4 | 11 to 12 | 4487 | CTCACTGTGTC ACCCAAACT | GGGAACCAGC TGCTCTGAAC | |||
| NEU1 ENST00000375631 | Sialidosis/Mucolipidosis I | 7647 | NEU1-1 | 1 to 6 | 5657 | CTGTGACTCATTCTC TCCACGACGACAGG | GAAGGTAGTGTCTGT CTCTCAAGCCTCCC |
| GNPTAB ENST00000299314 | Mucolipidosis II alpha/beta and Mucolipidosis III alpha/beta | 87,841 | GNPTAB-1 (Sanger) | 1 | 117 | CTATGCCCC TCCGTCCTC | CATACTGTATC GGGGCATCG |
| GNPTAB-2 (Sanger) | 2 | 86 | GTATGTGGTAG GCAGTAAGT | GTATATGTGCTG CTAAAGTG | |||
| GNPTAB-3 | 3 to 5 | 4816 | CCAAGACTACTCT ACTTCACCCACG | GCTCCACCTCCC AATACCATCATGC | |||
| GNPTAB-4 | 6 to 7 | 4732 | GAGACCAGGCCTC ACTCTGTCACCCA | CAGCCCTCTCCTC TGACATGCGACGT | |||
| GNPTAB-5 | 8 to 11 | 4809 | GAGTGGTGTGGACTT TCGTAGGGGTGGC | TGAGGGAGAGGGA AGAGCTGCTGAGGAG | |||
| GNPTAB-6 | 12 to 15 | 6862 | CTCTCCTCAGCA GCTCTTCCCTCTC | CTCCAGCTAGCCA CACCTGAAGTCC | |||
| GNPTAB-7 | 16 to 18 | 3542 | GGAAAAGAGCCAG ACCATACCTGCAT | GGGACCCTATCTCAA CTTGCAACTCCTATC | |||
| GNPTAB-8 | 19 to 21 | 9035 | CTCCCATAGCTAAA AGGCCATCTACCCTAG | CCACCCTACCTCTTC CTCTAACTGGTTGTA | |||
| GNPTG ENST00000204679 | Mucolipidosis III gamma | 13,391 | GNPTG-1 | 1 to 3 | 4209 | AACCCTGACCCG CTCTCCCCATCAC | CTCCCAGCCTGAC CCCTGCAACTCA |
| GNPTG-2 | 4 to 11 | 4268 | GTGCGTGGATAAT TGTGGTGTCTTGGC | GACGTGTTTCTCCC CGACCGTGGCTTT | |||
| MCOLN1 ENST00000264079 | Mucolipidosis Type IV | 13,783 | MCOLN1-1 | 1 to 7 | 6495 | GAATGTTGGAAGA CTCTGGGCTGGGG | GAAGGACAGGGA GCAGGTGAGGATGA |
| MCOLN1-2 | 7 to 14 | 6715 | CTGGTCAGGGAGT GTCTTGGGAGCA | GGATTAGTGGGTG GGGATGCGGGGT | |||
| GAA ENST00000302262.7 | Pompe disease | 19,524 | GAA-1 | 1 to 2 | 5610 | GATGAGGCAGCAG GTAGGACAGTGA | CTCTCAGGGCATA TCAGAAGAGGCG |
| GAA-2 | 3 to 14 | 8235 | CGTCAGGGAGTGG TCATGCAGAGAG | GTAACAGCACAGAG GAACGAGAGGC | |||
| GAA-3 | 15 to 20 | 4532 | GGAGCACCGTCAA CACTTAGCTAGG | TGACATGGGGAG GGTAGGTGAGGAG | |||
| HEXA ENST00000268097.9 | GM2 gangliosidosis - Tay-Sachs disease | 36,758 | HEXA-1 (Sanger) | 1 | 475 | TGATTCGCCGA TAAGTCACG | CTCGAGGAGG AAGTGGAGTG |
| HEXA-2 | 2 to7 | 7583 | GCCTCCTCTGCCT GATCCTTCTGTC | TGGCCCAGAGAC TACTTCCTGACGC | |||
| HEXA-3 | 8 to14 | 9518 | TACCCTCAGCCCT CCAGTGTGAGCC | GCTGGGACTACAA CCACACACCACCA | |||
| HEXB ENST00000511181.5 | GM2 gangliosidosis - Sandhoff disease | 83,825 | HEXB-1 (Sanger) | 1 | 907 | GGCAAAACCC TGTTTCGACA | GGTCCCTCCC AGATCCATTG |
| HEXB-2 | 2 to 9 | 10,013 | TTCCGCAACTGAG CACTTATAGGCC | GACTCTGACCAC ATGTTCACAGGCA | |||
| GALC ENST00000261304.6 | Krabbe disease | 143,640 | GALC-1 | 1 to 4 | 7760 | CGCCTCATCTCACAT AAGGGAAAACTCAGG | CAATGATCCTCCCAA TAGAACCTAAGCCCC |
| GALC-2 | 5 to 7 | 10,340 | GGGCTTAGGTTCTAT TGGGAGGATCATTGG | GGAAAGGAAGAGGA GTAGATAGACGCATGG | |||
| GALC-3 | 8 to 10 | 6698 | TTCTAAGGACAGGA GTAGTGGGGAGAGTAC | GACTACGATGAAGTG TAGATTCTGGGAGGG | |||
| GALC-4 | 11 to 14 | 7277 | GGTGTAGTTTGGTC AGTGTTCCATGTGC | GAGCCCTCTATGGT ATTCAAGTGCATGG | |||
| GALC-5 | 15 to 17 | 11,056 | CAGAGGACATACAA GAGAGACTGGGCTT | GATGCAGGTGAGGCTG TGGAGAAATAGAAG | |||
| GLA ENST00000218516.3 | Fabry disease | 11,323 | GLA | 1 to 7 | 10,756 | ACACATACACAG TCATGAGCGTCCAC | AGGTGGACAGGA AGTAGTAGTTGGCA |
| ASAH1 ENST00000637790.1 | Farber | 30,207 | ASAH-1 (Sanger) | 1 | 895 | CTCAACTGCT CCTTGTCCCT | TGCGAATCAC ACCCAGGTAT |
| ASAH1-2 | 2 to 4 | 6887 | CATGGAAGGGTGAGA GATGATAGGAAGTGC | CCCTAGGTGTTTCAT TGGTTCTGCGTCAAC | |||
| ASAH1-3 | 5 to 14 | 12,236 | GAGGGTGAATTCGTG CAGAGAGATAAGGAG | GGGTTTGCTGAGGAG GTAATCTAGGTCAAG |
PCR, polymerase chain reaction; LR-PCR, long-range PCR; MPS, mucopolysaccharidosis
Preparation of libraries and sequencing: PCR products of study participants were quantified on Qubit 3.0 fluorometer (Thermo Fisher Scientific, Massachusetts, USA) and pooled together at equimolar amounts. The samples were pooled to make a total volume of 100 μl with a final concentration of 1000 ng/100 μl (i.e. 10 ng/μl).
The study was performed in two phases. In the first phase, for standardization and validation of the study technique, DNA samples of the affected individuals and/or their parents with already identified gene variants (group A) were included, as listed in Table I. Four libraries were constructed in all to accommodate the 28 group A samples; each library consisted of 29 LR-PCR fragments of seven LSD genes, each fragment ranging from 5-10 kb in size. The analysis was performed in a blinded manner and the person analyzing the NGS data was not aware of the variants previously identified by Sanger sequencing, to eliminate any element of bias in the analysis.
In the second phase, samples of patients with enzymatically confirmed or biopsy proven diagnosis of specific LSDs and/or their parents, who had not already undergone mutation analysis (group B), were included, as shown in Table II. Five libraries were constructed to accommodate the 30 patient samples; each library consisted of LR-PCR fragments of 5 - 7 LSD genes, each ranging from 5 - 10 kb in size (24 fragments in libraries Lib_1 and Lib_3, 25 fragments in libraries Lib_2 and Lib_4 and 28 fragments in library Lib_5).
| Sample ID | Affected status | Library number | Name of the gene | LR-PCR fragment in which variant (s) identified | Sequence variant identified in the study (DNA notation) | Sequence variant identified in the study (protein notation) | Zygosity | Position of the variant | Known/Novel | Variant classification | Whether confirmed by Sanger sequencing |
|---|---|---|---|---|---|---|---|---|---|---|---|
| B_001 | Affected proband | Lib_1 | GBA | GBA-1 (Exons 1 to 11) | c.1448T>C | p.Leu483Pro | Hom | Exonic | Reported in ClinVar and HGMD | LP | Yes |
| B_002 | Affected proband | Lib_1 | GALNS | GALNS-5 (Exons 12 to 14) | c.1349A>G | p.Glu450Gly | Hom | Exonic | Reported in HGMD | VOUS | Yes |
| B_003 | Affected proband | Lib_1 | HEXA | HEXA-2 (Exons 2 to 7) | c.340G>A | p.Glu114Lys | Hom | Exonic | Reported in ClinVar | LP | Yes |
| B_004 | Unaffected carrier parent | Lib_1 | GLB1 | GLB1-2 (Exons 2 to 5) | c.522T>G | p.Tyr174Ter | Het | Exonic | Novel | P | Yes |
| B_005 | Unaffected carrier parent | Lib_1 | NPC1 | NPC1-8 (Exons 15 to 20) | c.2378_2378delA | p.Asn793Ile fsTer3 | Het | Exonic | Novel | LP | Yes |
| B_006 | Unaffected carrier parent | Lib_2 | GBA | GBA-1 (Exons 1 to 11) | c.492C>G | p.Ser164Arg | Het | Exonic | Novel | VOUS | Yes |
| B_007 | Affected proband | Lib_2 | GALNS | GALNS-1 (Exon 1) | c.120+1G>C | NA | Hom | Splice site | Reported in ClinVar | P | Yes |
| B_008 | Affected proband | Lib_2 | HEXA | HEXA-2 (Exons 2 to 7) | c.571-2A>G | NA | Hom | Splice site | Novel | LP | Yes |
| B_009 | Unaffected carrier parent | Lib_2 | GLB1 | GLB1-6 (Exons 13 to15) | c.1242delG | p.Phe415Leu fsTer46 | Het | Exonic | Novel | P | Yes |
| B_010 | Unaffected carrier parent | Lib_2 | NPC1 | NPC1-8 (Exons 15 to 20) | c.2800C>T | p.Arg934Ter | Het | Exonic | Reported in HGMD | P | Yes |
| B_011 | Affected proband | Lib_2 | NAGLU | NAGLU-1 (Exons 1to 6) | c.608G>C | p.Arg203Pro | Hom | Exonic | Novel | VOUS | Yes |
| B_012 | Affected proband | Lib_3 | GBA | GBA-1 (Exons 1 to 11) | c.538G>A | p.Asp180Asn | Hom | Exonic | Novel | VOUS | Yes |
| B_013 | Affected proband | Lib_3 | GALNS | GALNS-1 (Exon 1) | c.95A>C | p.Asn32Thr | Hom | Exonic | Novel | VOUS | Yes |
| B_014 | Affected proband | Lib_3 | HEXA | HEXA-3 (Exons 8 to14) | c.1424C>G | p.Pro475Arg | Hom | Exonic | Novel | VOUS | Yes |
| B_015 | Affected proband | Lib_3 | GLB1 | GLB1-7 (Exon-16) | c.1799C>T | p.Thr600Ile | Hom | Exonic | Novel | VOUS | Yes |
| B_016 | Affected proband | Lib_3 | NPC1 | NPC1-7 (Exons 12 to 14) | c.2050C>T | p.Leu684Phe | Hom | Exonic | Reported in ClinVar and HGMD | LP | Yes |
| B_017 | Affected proband | Lib_4 | GBA | GBA-1 (Exons 1 to 11) | c.1504C>T | p.Arg502Cys | Hom | Exonic | Reported in ClinVar and HGMD | P | Yes |
| B_018 | Affected proband | Lib_4 | GALNS | GALNS-2 (Exon 2 to 4) | c.376G>T | p.Glu126Ter | Hom | Exonic | Novel | P | Yes |
| B_019 | Affected proband | Lib_4 | GNPTAB | GNPTAB-8 (Exons 19 to 21) | c.3503_3504delTC | p.Leu1168Gln fsTer5 | Hom | Exonic | Reported in HGMD | P | Yes |
| B_020 | Unaffected carrier parent | Lib_4 | NPC1 | NPC1-8 (Exons 15 to 20) | c.2800C>T | p.Arg934Ter | Het | Exonic | Reported in HGMD | P | Yes |
| B_021 | Affected proband | Lib_4 | IDUA | IDUA-2 (Exon 3 to 14) | c.1853_1855delACC | p. 618_619del | Hom | Exonic | Reported in HGMD | LP | Yes |
| B_022 | Affected proband | Lib_4 | GNPTG | GNPTG-1 (Exon 1to 3) | c.111+1G>C | NA | Hom | Splice site | Novel | LP | Yes |
| B_023 | Affected proband | Lib_5 | GBA | GBA-1 (Exons 1 to 11) | c.492C>G/c. 254G>A | p.Ser164Arg/ p.Gly85Glu | CH | Exonic/Exonic | Novel/Reported in ClinVar and HGMD | VOUS/LP | Yes |
| B_024 | Unaffected carrier parent | Lib_5 | GALNS | GALNS-5 (Exon 12 to 14) | c.1483-1G>C | NA | Het | Splice site | Novel | P | Yes |
| B_025 | Affected proband | Lib_5 | NEU1 | NEU1-1 (Exons 1 to 6) | c.872T>C | p.Ile291Thr | Hom | Exonic | Novel | VOUS | Yes |
| B_026 | Affected proband | Lib_5 | IDS | NA | No significant variant identified | NA | NA | NA | NA | NA | NA |
| B_027 | Unaffected carrier parent | Lib_5 | GAA | GAA-3 (Exons 3 to 14) | c.1726G>A | p.Gly576Ser | Het | Exonic | Reported in ClinVar | LB | Yes |
| B_028 | Unaffected carrier parent | Lib_5 | GNPTAB | GNPTAB-3 (Exons 3 to 5) | c.1410+1 C>A | NA | Het | Splice site | Novel | LP | Yes |
| B_029 | Affected proband | Lib_5 | GLB1 | GLB1-7 (Exon-16) | c.1799C>T | p.Thr600Ile | Hom | Exonic | Novel | VOUS | Yes |
| B_030 | Affected proband | Lib_5 | IDUA | IDUA-2 (Exon 3 to 14) | c.1469T>C | p.Leu490Pro | Hom | Exonic | Reported in ClinVar and HGMD | LP | Yes |
Hom, homozygous; Het, heterozygous; CH, compound heterozygous; P, pathogenic; LP, likely pathogenic; VOUS, variant of uncertain significance; LB, likely benign; HGMD, human gene mutation database; PCR, polymerase chain reaction; LR-PCR, long-range PCR, NA, not avaialable
One sample for each gene was included in one library, as listed in Tables I and II. Equimolar long PCR amplicons corresponding to different genes of different patients (e.g. IDUA amplicons of one patient, ARSA amplicons of another patient and HEXA amplicons of a third patient) were combined and the pooled samples were subjected to sequence analysis using the Illumina MiSeq platform (Illumina Inc., San Diego, California, USA). Reads with 100X coverage for every amplicon were generated to ensure the accuracy of the results.
Data analysis: The Illumina sequencing reads in FASTQ format were checked for quality using the FastQC application (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Libraries for which data quality was found to be satisfactory were processed further for analysis. Paired-end reads were aligned, using the Burrows-Wheeler Aligner (BWA) version 0.5.9rc1, to the human genome assembly hg1910. Genome Analysis Toolkit (GATK) software package version 1.6 (Broad Institute, Cambridge, MA, USA)11 was used for variant calling12 and ANNOVAR software version 2012 was used for annotation of the variants13.
All the identified variants were filtered using the following criteria for each patient: (i) genetic filter - filtration of variants based on presence in known, mutation or polymorphism databases; (ii) evolutionary filter - filtration of variants based on evolutionary conservation in vertebrates; and (iii) functional filter: filtration of variants based on amino acid change, expression in the target tissue, computational prediction of deleterious effect, etc. The population databases 1000 Genome (https://www.internationalgenome.org/1000-genomes-browsers/) and gnomAD (https://gnomad.broadinstitute.org/) were used to filter out polymorphic variants with a high minor allele frequency. Previously reported known pathogenic variants were identified using the ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and HGMD (Human Gene Mutation Database; http://www.hgmd.cf.ac.uk/ac/index.php) databases and by searching published literature on PubMed. Classification of the identified variants was done as per the guidelines outlined by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP variant classification guidelines 2015)14. In silico prediction of the effect of the variants was done using mutation prediction tools such as PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (https://sift.bii.a-star.edu.sg/), MutationTaster2 (http://www.mutationtaster.org/) and CADD (https://cadd.gs.washington.edu/). The impact of the variants on splicing was checked with the help of the MutationTaster2 and Human Splicing Finder (http://www.umd.be/HSF/) software.
Sanger sequencing: The sequence variants identified through this study in the group B samples were validated by Sanger sequencing which was done on ABI 3130 Genetic Analyzer (Thermo Fisher Scientific, Massachusetts, USA) following the manufacturer’s protocol.
An overview of the workflow is shown in Figure 1.
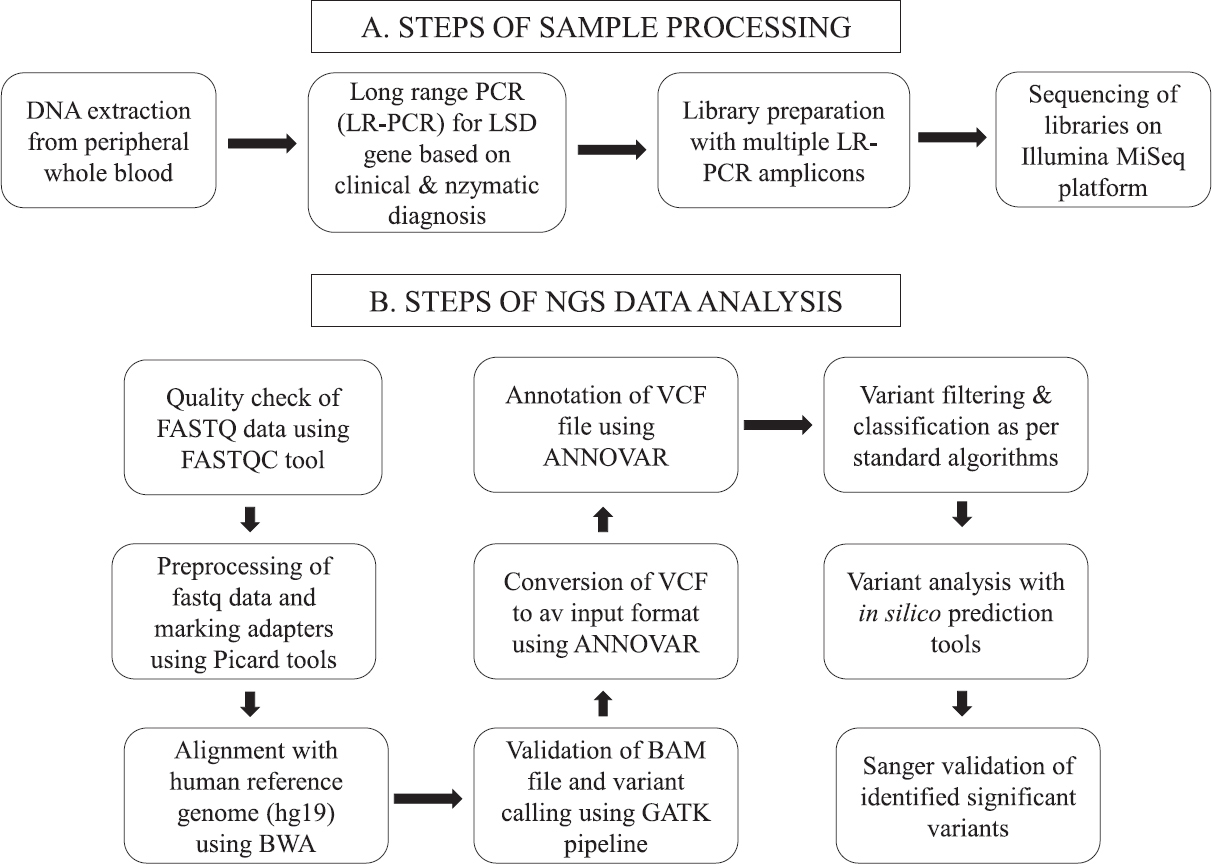
- Overview of the study workflow.
Initial standardization and validation of the study protocol were done using 28 samples of affected probands and/or carrier parents (group A) with known variants in seven genes, in the first phase of this study. Later, in the second phase, 30 samples of enzymatically confirmed or biopsy-proven patients with LSDs and/or their carrier parents who had not undergone any prior mutation analysis (group B) were included and the sequence variants identified in them through NGS were validated by targeted Sanger sequencing.
Results
Initially, for evaluating and validating the NGS workflow, 28 samples with previously identified variants (group A samples) were included in four libraries, as shown in Table I, and we found 100 per cent concordance in the sequence variants previously identified by Sanger sequencing and then by NGS in this study, ensuring that the analysis was done in a blinded manner to eliminate bias. Heterozygous variants in carrier parents, as well as biallelic variants (compound heterozygous or homozygous variants) in the probands, were identified with the NGS based study technique, as shown in Table I. Of the total 34 variants identified in the group A samples, 18 were missense variants, three were nonsense variants, eight were small indels and four were splice site variants; one variant identified in the ARSA gene (c.417C>T) is a synonymous variant and classified as ‘likely benign’ but was predicted to cause splice site changes and therefore to be disease-causing by MutationTaster2 (http://www.mutationtaster.org/).
In the second phase of the study, 30 samples of probands and/or their carrier parents were included. These DNA samples were collected from clinically diagnosed cases of different LSDs, where the diagnosis was confirmed by enzymatic analysis for all except those with NPC1 gene associated NPD type C (group B samples). There is no lysosomal enzyme assay available for NPD type C, as the NPC1 gene product is not an enzyme but a protein involved in intracellular cholesterol trafficking; therefore, these cases were chosen based on the clinical features of neonatal cholestasis with liver biopsy findings suggestive of NPD in the proband. Heterozygous as well as homozygous disease-causing variants were identified through the NGS workflow, as listed in Table II, and all these identified variants were validated by targeted Sanger sequencing. Figure 2 shows four representative examples of the variants identified through NGS and thereafter validated through Sanger sequencing. Of the total 30 variants identified in the group B samples, 17 were missense variants, four were nonsense variants, four were small indels and five were splice site variants. No significant variants were identified in the IDS gene (in any of the three LR-PCR fragments covering the IDS gene) in the sample of one patient clinically diagnosed and enzymatically confirmed to have mucopolysaccharidosis (MPS) type II (Hunter syndrome); Sanger sequencing of the exons and flanking intron-exon junctions of the IDS gene also did not reveal any significant variants. Further multiplex ligation-dependent probe amplification of the gene is planned for this patient to look for large exonic deletions/duplications and complex rearrangements, which are known to occur in around 20 per cent of patients with MPS II, due to recombination of the IDS gene with IDSP1 pseudogene. It is possible to detect large exonic deletions and duplications by analysis of NGS data using software which compare the read-depth of the test data with the matched aggregate reference dataset, but complex rearrangements may get missed by sequencing-based testing techniques.
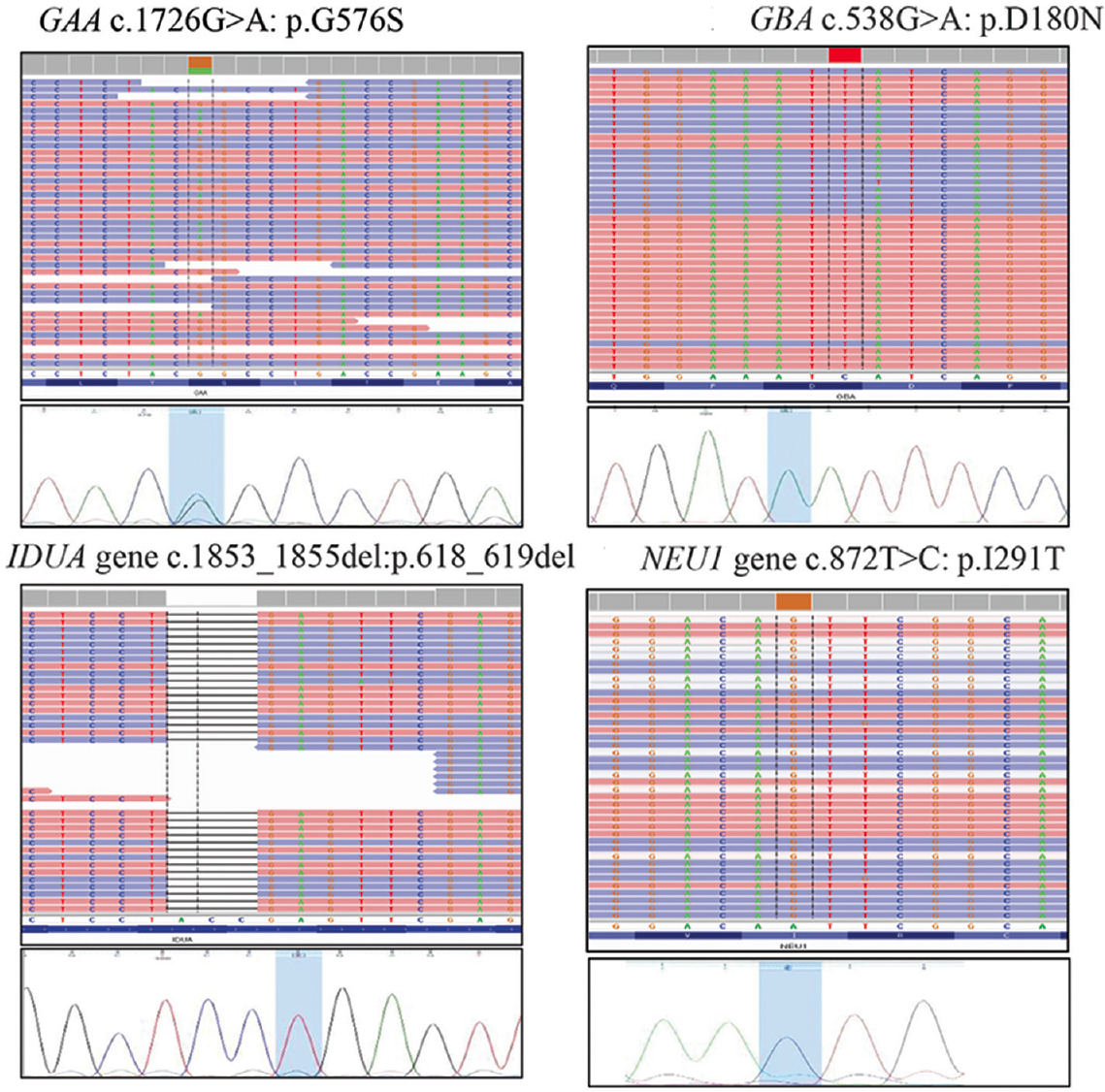
- Representative examples of the variants identified through the study protocol in four different samples and thereafter validated through targeted Sanger sequencing.
Discussion
The diagnostic journey for many patients with lysosomal storage disorders is often time-consuming, tedious and expensive, involving multiple enzyme assays and sometimes even invasive tests such as bone marrow and/or liver biopsy15. The availability of a multigene panel test for LSDs can make the diagnosis faster and cheaper and this has become possible with the availability of high throughput NGS technologies in recent years6,7,16.
Most of the commercially available kits for targeted ‘capture’ of genes for multigene panel tests are based on one of the following approaches: (i) hybridization-based strategies, (ii) transposon-mediated fragmentation (tagmentation), (iii) molecular inversion probes, and (iv) PCR based target enrichment9. However, these commercial capture kits are expensive, available in fixed quantities and are therefore not easily scalable, or readily customizable.
In this study, we have applied and validated a strategy based on the in-house development of selective amplicons through LR-PCR amplification for targeted capture of different LSD genes of interest, followed by NGS of pooled samples. The two most important advantages of using this method of amplicon libraries were the significant reduction in the costs and the ability to easily customize the genes of interest when compared to commercially available targeted enrichment kits. As has been shown in the study, this technique can be used for molecular genetic testing of already enzymatically confirmed cases, where molecular genetic testing can be done directly for the concerned gene only and it can also be used for targeted sequencing of already identified variants in family members. In addition, small panels can be custom designed based on the clinical phenotype of the patient for genes associated with overlapping phenotypes, e.g. MPS associated genes; the molecular diagnosis can be then validated by performing the concerned enzyme assay. The groups of genes included in each library can be flexible. Any combination of genes can be included in one library, as long as one ensures that there is no sequence homology between the different genes. Similarly, different patients’ samples can be included in one library, provided these are for different genes. Apart from these advantages, there are some additional benefits of this method. This technique has the ability to include complete intronic regions of genes provided the introns are not too large in size, as opposed to the commercially available kits that chiefly include the coding regions (exons) and the flanking intron-exon junctions only; thus, it helps to detect deep intronic variants also. Through appropriate designing of LR-PCR primers, one can also ensure that the gene is selectively amplified and pseudogene sequences, if any, are excluded. Table III lists the criteria that may be considered when selecting patients, clinically diagnosed to have various LSDs, for testing through this proposed method, and for preparing libraries of pooled samples.
| • Twenty five to 30 LR-PCR fragments, each of 5-10 kb size, can be included per library |
| • There should not be any sequence homology between the fragments |
| • LR-PCR fragments corresponding to different genes for one or more patients can be pooled together in one library |
| • Testing one or a few genes (up to 5) for a patient, makes the strategy most cost-effective; therefore, it is better to narrow down the diagnostic possibilities through prior clinical evaluation and laboratory testing |
| • Ideal for conditions where a specific gene has to be tested based on clinical and biochemical phenotype (e.g. ARSA for metachromatic leukodystrophy) or where a few genes (>1 but <5) have to be tested based on the clinical phenotype (e.g. MPS III or MPS IV) |
| • Ideal for genes such as GUSB (for MPS VII) and GBA (for Gaucher disease), which have pseudogenes, as LR-PCR primers can be suitably designed to selectively amplify the gene and exclude the pseudogene |
MPS, mucopolysaccharidosis; LR-PCR, long range-PCR
A similar approach was used by other researchers for designing multigene panels for groups of disorders such as Mendelian retinal disorders and hereditary breast cancer17-20. Their results also demonstrated that LR-PCR amplification, followed by NGS was an effective method for mutation analysis of monogenic heterogeneous diseases.
Analysis of the cost-effectiveness of this in-house LR-PCR amplification based targeted enrichment, followed by NGS based assay versus Sanger sequencing for individual genes as well as NGS based multigene panel sequencing following capture with commercially available kits revealed that this method was much more economical, especially for single genes which are within 10-50 kb in size and groups of small genes (especially less than 5). In this study, the approximate cost of sequencing for each LR-PCR fragment worked out to around 1000 Indian rupees [approximately 15 US dollars ($)]. Therefore, for a gene that can be covered with 1-5 LR-PCR fragments, the cost of sequencing by the study method would be only around 1000-5000 INR (₹) (around 15-75 US $). Thus, this testing strategy will be far more cost-effective than larger commercial panels such as clinical-exome sequencing (CES) and whole-exome sequencing (WES), if one has to look for variants in a single specific gene or a small number of genes based on the clinical suspicion. For example, if the clinical diagnosis is mucopolysaccharidosis type III (MPS III), one can do sequencing only for SGSH (for MPS IIIA) and NAGLU (for MPS IIIB), as MPS IIIA and IIIB together account for around 90 per cent of MPS III. The cost for SGSH and NAGLU mutation analysis, with the proposed LR-PCR amplification, followed by the NGS method, would come to only around ₹ 3000 (approximately 40 US $), which is significantly less than the cost of CES and WES.
Despite its cost-effectiveness, robustness and easy customizability, an assay based on this model has some limitations. One is the requirement to pool samples and do the testing in batches, to maintain cost-effectiveness, which makes the method unsuitable when a sample has to be tested within a short and limited time frame, for instance, for a prenatal sample, where the report has to be issued as early as possible. The other important drawback of this strategy is that it is not suitable for panel testing of larger numbers of genes, as doing LR-PCR for multiple gene fragments would be tedious and time consuming. Furthermore, when designing libraries, one has to ensure that more than one patient’s sample for the same gene is not included and there is no sequence homology between the different genes included in one library.
This study has demonstrated the utility of the technique of LR-PCR amplification, followed by NGS in the molecular diagnosis of individuals with lysosomal storage disorders. In particular, the cost-effectiveness of this method has been demonstrated in the Indian context. The relatively low sample size and the inclusion of only 22 LSD associated genes are some of the limitations of this study.
Overall, LR PCR-based targeted gene enrichment combined with NGS appears to be a reliable, clinically practical, easily customizable, scalable and cost-effective approach for mutation analysis of lysosomal storage disorders and can be used even in resource-poor settings. We plan to apply the same strategy to develop molecular genetic assays for other groups of disorders with overlapping phenotypes such as organic acidurias, other inborn errors of metabolism, coagulopathies, pathway disorders, etc. especially where functional assays are available to identify the gene to be sequenced or to validate the molecular study results.
Financial support and sponsorship
The molecular genetic tests in this study were performed with the help of funding support obtained under Department of Health Research, Indian Council of Medical Research, New Delhi (GIA/62/2014-DHR).
Conflicts of interest
None.
References
- Lysosomal disorders. In: The metabolic and molecular bases of inherited disease (8th ed). New York: McGraw-Hill; 2001. p. :3371-877.
- Lysosomal storage disorders: Molecular basis and laboratory testing. Hum Genomics. 2011;5:156-69.
- [Google Scholar]
- Epidemiology of lysosomal storage diseases: An overview. In: Mehta A, Beck M, Sunder-Plassmann G, eds. Fabry disease: Perspectives from 5 years of FOS, Ch. 2. Oxford: Oxford PharmaGenesis; 2006.
- [Google Scholar]
- Measurement of lysosomal enzyme activities: A technical standard of the American College of Medical Genetics and Genomics (ACMG) Genet Med. 2022;24:769-83.
- [Google Scholar]
- Prenatal diagnosis of Pompe disease: Enzyme assay or molecular testing? Indian Pediatr. 2011;48:901-2.
- [Google Scholar]
- Setup and validation of a targeted next-generation sequencing approach for the diagnosis of lysosomal storage disorders. J Mol Diagn. 2020;22:488-502.
- [Google Scholar]
- Sensitivity, advantages, limitations, and clinical utility of targeted next-generation sequencing panels for the diagnosis of selected lysosomal storage disorders. Genet Mol Biol. 2019;42((1 Suppl 1)):197-206.
- [Google Scholar]
- Overview of target enrichment strategies. Curr Protoc Mol Biol. 2015;112:7.21.1-23.
- [Google Scholar]
- Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754-60.
- [Google Scholar]
- The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297-303.
- [Google Scholar]
- A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491-8.
- [Google Scholar]
- ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
- [Google Scholar]
- Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24.
- [Google Scholar]
- Current molecular genetics strategies for the diagnosis of lysosomal storage disorders. Expert Rev Mol Diagn. 2016;16:113-23.
- [Google Scholar]
- Inherited metabolic disorders: Efficacy of enzyme assays on dried blood spots for the diagnosis of lysosomal storage disorders. JIMD Rep. 2017;31:15-27.
- [Google Scholar]
- Detection of genomic variations in BRCA1 and BRCA2 genes by long-range PCR and next-generation sequencing. J Mol Diagn. 2012;14:286-93.
- [Google Scholar]
- Detection of novel mutations that cause autosomal dominant retinitis pigmentosa in candidate genes by long-range PCR amplification and next-generation sequencing. Mol Vis. 2013;19:654-64.
- [Google Scholar]
- Long-range PCR in next-generation sequencing: Comparison of six enzymes and evaluation on the MiSeq sequencer. Sci Rep. 2014;4:5737.
- [Google Scholar]
- Long-range PCR-based NGS applications to diagnose mendelian retinal diseases. Int J Mol Sci. 2021;22:1508.
- [Google Scholar]




