
Translate this page into:
Cystic fibrosis transmembrane conductance regulator-related male infertility: Relevance of genetic testing & counselling in Indian population
For correspondence: Dr Rahul Gajbhiye, Department of Clinical Research, National Institute for Research in Reproductive Health, Indian Council of Medical Research, J.M. Street, Parel, Mumbai 400 012, Maharashtra, India e-mail: gajbhiyer@nirrh.res.in
-
Received: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
Due to limited information available on the frequency and spectrum of cystic fibrosis (CF) transmembrane conductance regulator (CFTR) gene mutations in congenital bilateral absence of vas deferens (CBAVD) in Indian population, it is difficult to provide accurate genetic counselling to couples. The present study was undertaken to investigate the spectrum and frequency of CFTR gene mutations in Indian men with CBAVD and to determine the female CF carrier status.
Methods:
Direct DNA sequencing of the CFTR gene was carried out in eighty CBAVD men, their female partners and fifty controls from the general population. Pathological significance of the identified novel CFTR gene variants was carried out using in silico tools. Appropriate genetic counselling was provided to the couples prior to intracytoplasmic sperm injection (ICSI).
Results:
A significant association was observed for CFTR gene variants in Indian CBAVD men versus controls (odds ratio: 12.1; 95% confidence interval: 4.8-30.4; P<0.0001). A total of 20 CFTR gene variants were identified in 53 CBAVD men. Eight novel missense CFTR gene variants (L214V, A238P, E379V, L578I, F587L, L926W, R1325K and R1453Q); two novel splice-site gene variants (c.1-30C>G and IVS1+2T>G) and ten previously reported mutations (R75Q, c.1210-12[5], F508del, A309G, R334W, I444T, R668C, R709X, A1285V and Q1352H) were detected in CBAVD men. The novel and reported CFTR gene mutations were L926W (2.5%, P=0.26), R1453Q (2.5%, P=0.26), F508del (8.75%, P=0.03) and c.1210-12[5] (42.5%, P=0.002). A total of 13 (16.2%) female partners were found to be a CF carrier. Nine couples had a risk of transmitting mutant CFTR allele to the offspring.
Interpretation & conclusions:
The heterogeneous spectrum of CFTR gene in Indian population suggests the necessity of screening CBAVD men and female partners for accurate genetic counselling prior to undergoing ICSI.
Keywords
Azoospermia
cystic fibrosis transmembrane conductance regulator gene
congenital bilateral absence of vas deferens
genetic counselling
intracytoplasmic sperm injection
male infertility

Congenital bilateral absence of vas deferens (CBAVD), an autosomal recessive disorder, accounts for 1-2 per cent of male infertility with an estimated incidence of 1:1000 men1. CBAVD is considered a primary genital form of cystic fibrosis (CF), associated with abnormalities in the CF transmembrane conductance regulator (CFTR) gene2. In humans, the CFTR gene on chromosome 7q31.2 encodes CFTR protein that regulates the chloride ions across epithelial cell membranes2. Structurally, the protein comprises: two membrane-spanning domains which form the channel pores, two nucleotide-binding domains which control channel gating and one regulatory domain (R domain) which determines the phosphorylation activity3.
More than 2000 CFTR gene variants have been documented in the CF mutations Databases (http://www.genet.sickkids.on.ca/cftr/app, http://www.umd.be/CFTR, http://www.cftr2.org/). F508del and the c.1210-12[5] are also referred as (5T) the most common severe and mild mutations, respectively, detected in CF- and CFTR-related disorders (CFTR-RDs) such as CBAVD1. There is a variation in the frequency of F508del, while the 5T variant is found to be present at a similar frequency in CBAVD men with different ethnicity and residing in different geographical regions1234567891011121314. In North Indian CBAVD men, the frequency of F508del and c.1210-12[5] was reported as 11-34 and 26-54 per cent, respectively459. Our previous study reported 39.4 per cent frequency of IVS9-c.1210-12[5] in CBAVD men7. Currently, there is no epidemiological statistics on the incidence of CBAVD phenotype in India. However, data accumulated over a decade suggest that the incidence of isolated CBAVD in Indian population might be comparable to that of Caucasians456789.
CBAVD men have normal spermatogenesis and can achieve biological paternity using assisted reproductive technologies (ART) such as intracytoplasmic sperm injection (ICSI)15 with their own sperm retrieved through various sperm retrieval techniques such as microsurgical epididymal sperm aspiration, testicular sperm extraction, percutaneous epididymal sperm aspiration16 or opt for donor insemination. Moreover, if the female partner of a CBAVD male is a CF carrier, she possesses a risk of transmitting the mutant CFTR allele to the progeny, resulting in a child born with a full-blown CF or CF-related disorders such as CBAVD1617, highlighting the significance of screening female partners. However, CF carrier frequency in the female partner of CBAVD men still remains undetermined. Routine screening for CFTR gene is not conducted in ART clinics in India due to inadequate knowledge of the frequency and spectrum of CFTR gene variants, limited awareness of the CFTR-related male infertility and non-availability of population-specific mutation panel.
Therefore, the present study was undertaken to identify the spectrum and frequency of CFTR gene variants in Indian CBAVD men and to determine their female partner CF carrier status. It was also aimed to understand the genetic risks involved, if the female partner was a CF carrier and to provide accurate counselling to the couples prior to undergoing ICSI.
Material & Methods
This study was approved by the Institutional Ethics Committee for research involving human subjects at ICMR-National Institute for Research in Reproductive Health (ICMR-NIRRH), Mumbai, India. Written informed consent was also obtained from all the study participants prior to blood sample collection.
The present study represents a part of the case–control genetic study conducted during the period 2011-2017 at ICMR-NIRRH, Mumbai. The total sample size was calculated as CBAVD (n=100), female partners of CBAVD (n=100) and fertile controls (n=50). A total of eighty unrelated sub-fertile men with a confirmed diagnosis of CBAVD were recruited from different geographic regions in India [north (n=16), northeast (n=1), west (n=48), central (n=1) and south (n=14)]. The semen parameters were evaluated as reported previously18. Men with semen parameters showing azoospermia, low seminal volume (<1.5 ml), absence/low fructose in semen and absence of the bilateral vas deferens on scrotal examination by the andrologists (Drs Rupin Shah and Vijay Kulkarni) were recruited as CBAVD cases. Testicular volume was assessed using Prader Orchidometer (Holtain Ltd, Crosswell, Wales, UK). Ultrasonography (USG) of the abdomen and pelvis was also performed. CBAVD men with renal abnormalities or those with congenital unilateral absence of vas deferens were excluded from the data analysis. Transrectal ultrasonography (TRUS) was carried out in CBAVD men to detect anomalies of seminal vesicles and ejaculatory ducts, wherever possible. Female partners of CBAVD men (n=80) were recruited in this study to assess carrier frequency. Proven fertile men from the general population attending the Family Welfare Clinics at ICMR-NIRRH were recruited as controls (n=50). The controls had normal semen parameters with bilaterally palpable vas deferens. Study participants having family or personal history of CF or CF-related disorders were excluded from the study. The information related to ICSI outcomes were obtained during the follow up of the CBAVD men with the andrologist.
Cystic fibrosis transmembrane conductance regulator (CFTR) gene testing and analysis: Peripheral venous blood was collected through venipuncture in ethylenediaminetetraacetic acid (EDTA) tubes, and genomic DNA was isolated using QIAamp®DNA Blood Midi Kit (Qiagen Inc., Hilden, Germany). The essential promoter, entire coding regions and splice sites of 27 exons of the CFTR gene were amplified by PCR using specific primers (Supplementary Table I). Sequencing reaction and analysis were carried out as described earlier6.
| Amplification site | PCR primer | Amplicon length (bp) | Annealing temperature (ºC/35 cycles) |
|---|---|---|---|
| Promoter + Exon 1 | Forward: 5’ - GTGGAGAAAGCCGCTAGAGCAAAT - 3’ Reverse: 5’- GCAAGTAGATGTGGCTCTCTA - 3’ |
360 | 58 |
| Exon 2 | Forward: 5’ - GTGCATAATTTTCCATATGCC - 3’ Reverse: 5’- TTAGCCACCATACTTGGCTC- 3’ |
341 | 58 |
| Exon 3 | Forward: 5’ - CATGCAACTTATTGGTCCCAC - 3’ Reverse: 5’- TTCACCAGATTTCGTAGTCTTTTC - 3’ |
232 | 58 |
| Exon 4 | Forward: 5’ - TCTTGTGTTGAAATTCTCAGGG - 3’ Reverse: 5’- AAAACTACAACAGAGGCAGTTTACAG - 3’ |
525 | 58 |
| Exon 5 | Forward: 5’ - GAACCTGAGAAGATAGTAAGCTAGATG - 3’ Reverse: 5’- GAAAACTCCGCCTTTCCAG - 3’ |
321 | 56 |
| Exon 6 | Forward: 5’ - GGGTGGAAGATACAATGACACC - 3’ Reverse: 5’- TCCTGGTTTTACTAAAGTGGGC - 3’ |
287 | 56 |
| Exon 7 | Forward: 5’ - TGCCCATCTGTTGAATAAAAG - 3’ Reverse: 5’- CAAACATCAAATATGAGGTGGAAG- 3’ |
340 | 57 |
| Exon 8 | Forward: 5’ - CTTCCATTCCAAGATCCCTG - 3’ Reverse: 5’- TGAACATTCCTAGTATTAGCTGGC- 3’ |
476 | 57 |
| Exon 9 | Forward: 5’ - TGCTTGGCAAATTAACTTTAGAAC - 3 Reverse: 5’- GCACCTGGCCATTCCTCTAC - 3’ |
440 | 58 |
| Exon 10 | Forward: 5’ - GGCCATGTGCTTTTCAAACT - 3’ Reverse: 5’- CGCCAACAACTGTCCTCTTT - 3’ |
270 | 56 |
| Exon 11 | Forward: 5’ - CAAGTGAATCCTGAGCGTGA - 3’ Reverse: 5’- CCGATTGAATATGGAGCCAA - 3’ |
336 | 58 |
| Exon 12 | Forward: 5’ - GGAAGATGTGCCTTTCAAATTC - 3’ Reverse: 5’- CAGCAAATGCTTGCTAGACC - 3’ |
204 | 58 |
| Exon 13 | Forward: 5’ - GACCAGGAAATAGAGAGGAAATG - 3’ Reverse: 5’- TGCAATCTATGATGGGACAG - 3’ |
213 | 56 |
| Exon 14 | Forward: 5’ - AAATGCTAAAATACGAGACATATTGC - 3’ Reverse: 5’- GGATGCTGTTGTCTTTCGGT - 3’ |
678 | 58 |
| Exon 15 | Forward: 5’ - ACAATGGTGGCATGAAACTG - 3’ Reverse: 5’- TGAGCTTTCGAATCTCTTAACC - 3’ |
547 | 57 |
| Exon 16 | Forward: 5’ - AATTTAGATGTGGGCATGGG - 3’ Reverse: 5’- GGATTACAATACATACAAACATAGTGG - 3’ |
201 | 58 |
| Exon 17 | Forward: 5’ - GGCTGCCAAATAACGATTTC - 3’ Reverse: 5’- GATGGTGGATCAGCAGTTTC - 3’ |
329 | 58 |
| Exon 18 | Forward: 5’ - GAGAAATTGGTCGTTACTTGAATC - 3’ Reverse: 5’- CTAAATGTGGGATTGCCTCAG - 3’ |
566 | 58 |
| Exon 19 | Forward: 5’ - AAATGTTTACTCACCAACATGTTTTC - 3’ Reverse: 5’- TTTACAAGATGAGTATCGCACATTC - 3’ |
225 | 62 |
| Intron 19 | Forward: 5’ - CATTCAGTGGGTATAAGC AGC - 3’ | 327 | 58 |
| Exon 20 | Forward: 5’ - TCTATTCAAAGAATGGCACCAG - 3’ Reverse: 5’- CAATGGAAATTCAAAGAAATCAC- 3’ |
549 | 56 |
| Exon 21 | Forward: 5’ - CATGAGGTTCATTTACGTCTTTTG - 3’ Reverse: 5’- CACAGTGACCCTCAATTTATCTG - 3’ |
276 | 57 |
| Exon 22 | Forward: 5’ - TTGTGAAATTGTCTGCCATTC - 3’ Reverse: 5’- CACAGTCTAACAAAGCAAGCAG - 3’ |
339 | 57 |
| Exon 23 | Forward: 5’ - CTGAATTATGTTTATGGCATGG - 3’ Reverse: 5’- TTGCAGAGTAATATGAATTTCTTGAG - 3’ |
317 | 56 |
| Exon 24 | Forward: 5’ - TGATGGTAAGTACATGGGTGTTTC - 3’ Reverse: 5’- TCAGCCATTTGTGTTGGTATG - 3’ |
283 | 62 |
| Exon 25 | Forward: 5’ - TCAAATGGTGGCAGGTAGTG - 3’ Reverse: 5’- TGATTCTGTTCCCACTGTGC - 3’ |
362 | 58 |
| Exon 26 | Forward: 5’ - CTACCCCATGGTTGAAAAGC - 3’ Reverse: 5’- TGAGTAAAGCTGGATGGCTG - 3’ |
421 | 58 |
| Exon 27 | Forward: 5’ - GTCTGACCTGCCTTCTGTCC - 3’ Reverse: 5’- ATTCCATGAGCAAATGTCCC - 3’ |
291 | 58 |
PCR, polymerase chain reaction
In silico prediction of pathogenicity: Bioinformatics tools such as PolyPhen-2, SIFT and Mutation Taster were used to predict the pathological significance of the identified novel CFTR variants. Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) predicted the possible impact of an amino acid substitution on the structure and function of a protein using physical and comparative considerations. Sorting Intolerant From Tolerant (SIFT) (https://sift.bii.a-star.edu.sg) and Mutation Taster (http://www.mutationtaster.org) predicted the effect of amino acid substitution on protein function based on sequence homology and the physical properties of amino acids. The pathological splicing effects of the exonic and intronic variants were predicted using Human Splicing Finder version 3.0 (http://www.umd.be/HSF3/HSF.html). Multiple sequence alignments using Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/) were carried out for understanding the conserved nature of the novel CFTR variants across various species.
Statistical analysis: All data were expressed as mean ± standard deviation (SD). Differences between proportions of CFTR gene mutations were compared by Chi-square test using STATA software (version 8.2, Texas, USA), and P<0.05 was considered statistically significant.
Results
Demographic and clinical characteristics: The age of CBAVD men was 31.97±5.18 yr (mean ± SD), range: 23-47 yr. The mean age of the female partners of CBAVD men was 27.78±4.61 yr, (range: 20-38 yr). The duration of infertility was 4.45±3.48 yr, (range: 12 to 240 months). The mean testicular size found was 20.6±4.5 ml. Based on the TRUS reports available, agenesis of seminal vesicles was found in 15 (18.8%) CBAVD men. The head of the epididymis was palpable in most of the CBAVD men. The semen parameters demonstrated azoospermia with a mean semen volume of 0.79±0.9 ml and a mean semen pH of 6.72±0.8.
Genotypic characteristics: Screening of complete CFTR gene in CBAVD men led to the identification of twenty variants comprising ten novel and ten known variants, as represented in Table I. The variants detected comprised 16 missense, two splicing, one non-sense and one frame shift mutations. Out of 80 CBAVD men, CFTR gene mutations were detected in 53 CBAVD men with 66.3 per cent frequency (Table II). A statistically significant association was observed for CFTR gene variants between Indian CBAVD men and controls (odds ratio: 12.1; 95% confidence interval: 4.8-30.4; P<0.0001).
| Mutations | Nucleotide change | Consequences | Exon/intron | Number of alleles |
|---|---|---|---|---|
| c.1-30C>G* | Splice error | - | 5’ UTR region | 1 |
| IVS1+2T>G* | c.53+2T>G | - | Intron 1 | 2 |
| R75Q | c.224G>A | Arginine to glutamine at 75 | Exon 3 | 3 |
| L214V* | c.640C>G | Leucine to valine at 214 | Exon 7 | 1 |
| A238P* | c.844G>C | Alanine to proline at 238 | Exon 7 | 1 |
| A309G | c.926C>G | Alanine to glycine at 309 | Exon 7 | 1 |
| R334W | c.1000C>T | Arginine to tryptophan at 334 | Exon 8 | 1 |
| 5T | c.1210-12[5] | Aberrant splicing | Intron 9 | 34 |
| E379V* | c.1136A>T | Glutamic acid to valine at 379 | Exon 9 | 1 |
| F508del | c.1521_1523delCTT | Deletion of phenylalanine at 508 | Exon 11 | 7 |
| I444T | c.1331T>C | Isoleucine to threonine at 444 | Exon 9 | 1 |
| L578I* | c.1732C>A | Leucine to isoleucine at 578 | Exon 13 | 1 |
| F587L* | c.1762T>G | Phenylalanine to leucine at 587 | Exon 13 | 2 |
| R668C | c.2002C>T | Arginine to cysteine at 668 | Exon 14 | 1 |
| R709X | c.2125C>T | Arginine to pre-mature stop codon at 709 | Exon 14 | 2 |
| L926W* | c.2777T>G | Leucine to tryptophan at 926 | Exon 17 | 2 |
| A1285V | c.3854C>T | Alanine to valine at 1285 | Exon 20 | 1 |
| R1325K* | c.3974G>A | Arginine to lysine at 1325 | Exon 25 | 1 |
| Q1352H | c.4056G>C | Glutamine to histidine at 1352 | Exon 25 | 2 |
| R1453Q* | c.4358G>A | Arginine to glutamine at 1453 | Exon 27 | 2 |
*Novel sequence variant. UTR, untranslated region
| Genotype | CBAVD men (n=80), n (%) | Female partner of CBAVD men (n=80), n (%) | Fertile men as controls (n=50), n (%) |
|---|---|---|---|
| CFTR variant detected | 53/80 (66.25)* | 13/80 (16.25)* | 7/50 (14) |
| At least one severe/mild variant | 41/80 (51.25)* | 13/80 (16.25) | 7/50 (14) |
| Two or more variants | 12/80 (15)* | 0/80 (0) | 0/50 (0) |
*P≤0.0001 as compared to controls. CBAVD, congenital bilateral absence of vas deferens
Novel variants: Of the ten novel variants, seven were heterozygous missense [L214V (KU325495), A238P (KT121468), E379V (KM596832), L578I (KU325496), L926W (KM014732), R1325K (KM596834) and R1453Q (KM596833)]. A homozygous missense gene variant F587L (KJ606925) was also identified. Electropherograms of the novel CFTR gene variants are represented in Fig. 1. Two of the novel splice-site variants detected were c.1-30C>G (KU325497) in the essential promoter region and IVS1+2T>G (KU325498). The most frequently detected novel gene variants were L926W (2.5%) and R1453Q (2.5%). The correlation of novel CFTR gene variants with semen characteristics, testicular size and epididymal status is shown in Table III.
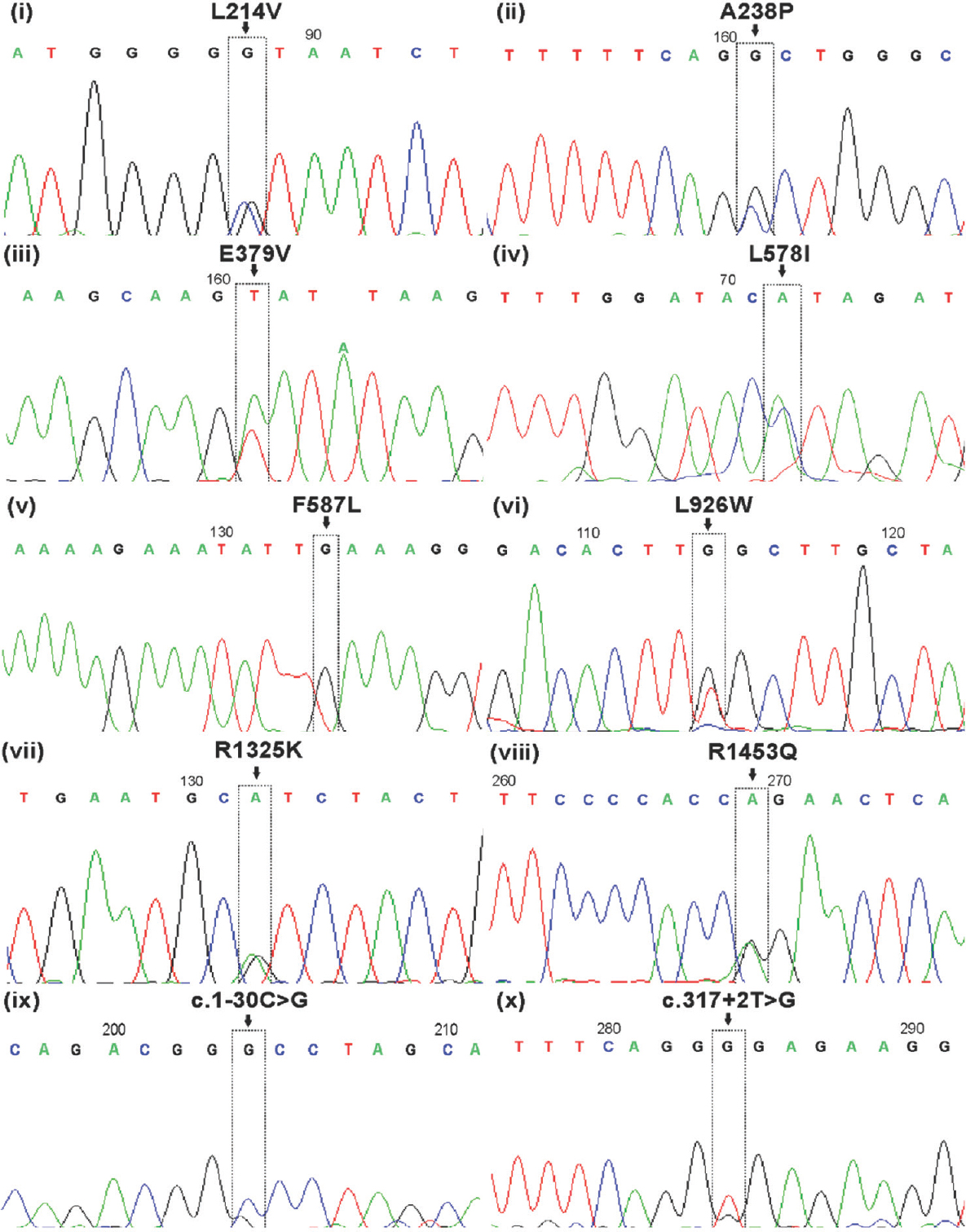
- Sequencing results of novel CFTR gene variants (L214V, A238P, E379V, L578I, F587L, L926W, R1325K, R1453Q, c.1-30C>G and c.317+2T>G) identified in Indian congenital bilateral absence of vas deferens men.
| CBAVD case number and geographical location | Semen parameters | Testicular size (ml) | Epididymal status | Novel CFTR gene variant | |||||
|---|---|---|---|---|---|---|---|---|---|
| Volume (ml) | PH | Liquefaction time (min) | Fructose | Right | Left | Right | Left | ||
| H4 (Maharashtra) | 0.5 | 6 | 20 | Negative | 30 | 30 | Head turgid | Head turgid | L926W |
| H20 (Maharashtra) | 0.3 | 6 | ND | Negative | 18 | 18 | Half head full | Half head full | c.1-30C>G |
| H21 (Maharashtra) | 1 | 8 | 60 | Negative | 18 | 15 | Head | Half head full | IVS1+2T>G, L926W |
| H22 (Uttar Pradesh) | 0.3 | 6 | 15 | Negative | 24 | 26 | Head turgid | Cystic head | R1453Q |
| H23 (Kerala) | 1 | 7.8 | ND | Negative | 22 | 22 | Head | Head | L578I |
| H28 (Uttar Pradesh) | 2 | 7.2 | ND | Negative | 30 | 30 | Head turgid | Head turgid | IVS1+2T>G |
| H29 (Andhra Pradesh) | 1.5 | 7.2 | 40 | Negative | 25 | 25 | Head | Head | R1325K |
| H31 (Uttar Pradesh) | 0.1 | 6 | 30 | Negative | 20 | 20 | Head | Head | R1453Q |
| H39 (Maharashtra) | 1 | 7.2 | 15 | Positive | 15 | 15 | One third head | One third head | F587L |
| H55 (Maharashtra) | 0.5 | 6 | 20 | Negative | 20 | 20 | Head | Head | E379V |
| H60 (Maharashtra) | 1 | 7 | 30 | Negative | 15 | 15 | Head | Head | A238P |
| H70 (Karnataka) | 0.1 | 6 | 20 | Negative | 16 | 14 | Head | Head | L214V |
ND, not done; CBAVD, congenital bilateral absence of vas deferens
Previously reported mutations: Ten previously known CFTR gene mutations identified in the study population are represented in Table I. Electropherograms of previously reported CFTR gene variants are represented in Fig. 2. The mutations/variants identified spanned the different domains of the CFTR protein and are represented in Fig. 3. The geographic distribution of CFTR gene variants in Indian CBAVD men is depicted in

- Sequencing results of previously reported CFTR gene mutations (F508del, R75Q, A309Q, R334W, I144T, R668C, R709X, A1285V and Q1352H) identified in Indian congenital bilateral absence of vas deferens men.

- Schematic diagram representing putative domain-type structure of the CFTR protein and identified CFTR gene variants. Novel CFTR gene variants are indicated in red and *. Previously known CFTR gene mutations are indicated in black. MSD, membrane-spanning domain; NBD, nucleotide-binding domain; R domain, regulatory domain.
Coding single-nucleotide polymorphisms and intronic variants: Of the 26 intronic variants detected, 19 were novel (Supplementary Table II), while 7 were previously reported. Four potential regulatory coding single-nucleotide polymorphisms namely M470V (n=55), T854T (n=21), P1290P (n=4) and Q1463Q (n=30) were detected in CBAVD men.
| Novel intronic variants | Intron | Interpretation |
|---|---|---|
| c.185+84G>A | 1 | Alteration of an intronic ESS site Creation of an intronic ESE site |
| c.405+24A>T | 3 | No significant splicing motif alteration detected |
| c.621+91G>A | 4 | Alteration of an intronic ESS site |
| c.875+12G>A | 6 | No significant splicing motif alteration detected |
| c.1248+80A>C | 8 | Creation of an intronic ESE site |
| c.1249-106A>T | 9 | Alteration of an exonic ESE site |
| c.1259-167A>C | 9 | No significant splicing motif alteration detected |
| c.1898+45A>T | 13 | No significant splicing motif alteration detected |
| c.1898+25delT | 13 | No significant splicing motif alteration detected |
| c.2751+855delTA | 15 | Alteration of an intronic ESS site Creation of an intronic ESE site |
| c.2751+106T>A | 15 | Alteration of an intronic cryptic acceptor site Creation of an intronic ESE site |
| c.2751+174A>C | 15 | Creation of an intronic ESE site |
| c.2751+86T>C | 15 | No significant splicing motif alteration detected |
| c.2751-75T>G | 15 | Activation of an intronic cryptic acceptor site |
| c.3120+529insC | 19 | No significant splicing motif alteration detected |
| c.3120+228T>C | 19 | No significant splicing motif alteration detected |
| c.4095+61T>A | 24 | Alteration of an intronic ESS site Creation of an intronic ESE site |
| c.4268+30G>A | 25 | No significant splicing motif alteration detected |
| c.4269-93A>T | 26 | Alteration of an exonic ESE site |
ESS, exonic splicing silencer; ESE, exonic splicing enhancer
Significant differences were observed in the frequency of CFTR gene variants across the isolated CBAVD and CBAVD with agenesis of seminal vesicle (P<0.001) Supplementary Table III.
| Mutations/novel variants | Isolated CBAVD (n=130 alleles) | CBAVD with agenesis of seminal vesicle (n=30 alleles) |
|---|---|---|
| c.1-30C>G* | 1 | 0 |
| IVS1+2T>G* | 2 | 0 |
| R75Q | 3 | 0 |
| L214V* | 1 | 0 |
| A238P* | 1 | 0 |
| A309G | 1 | 0 |
| R334W | 1 | 0 |
| 5T [c.1210-12T[5]] | 30 | 4 |
| E379V* | 1 | 0 |
| F508del | 7 | 0 |
| I444T | 1 | 0 |
| L578I* | 1 | 0 |
| F587L* | 2 | 0 |
| R668C | 1 | 0 |
| R709X | 2 | 0 |
| L926W* | 2 | 0 |
| A1285V | 1 | 0 |
| R1325K* | 1 | 0 |
| Q1352H | 1 | 1 |
| R1453Q* | 2 | 0 |
*Novel sequence variant. CBAVD, congenital bilateral absence of vas deferens
Female partner cystic fibrosis carrier status: Of the eighty female partners of the CBAVD men screened, CFTR gene variant was detected in 13 (16.2%) females (Table II). Twelve females harboured a c.1210-12[5] allele, while one was a carrier of A1285V heterozygous missense CFTR gene variant.
In silico analysis: In silico analysis of the CFTR gene variants is represented in Supplementary Table IV. PolyPhen-2 predicted six novel CFTR gene sequence variants to have a score >0.5 which is the threshold for damaging mutations and therefore, it might be responsible in damaging the CFTR protein structure or function. Similarly, SIFT predicted six novel variants to be damaging, whereas Mutation Taster predicted seven variants to be disease-causing mutations. In silico analysis using Human Splicing Finder v.3.0 predicted novel exonic variants to affect the splicing mechanism, either by altering or creating Exonic splicing silencer and Exonic splicing enhancer sites in the CFTR gene (Supplementary Table IV). Multiple sequence alignments using Clustal Omega confirmed the wild-type amino acid sequence of all the novel variants except R1325K to be conserved across species such as human, mouse, rabbit, bovine, sheep, monkey, pig and horse (
| Novel variants | Nucleotide change (HGVS nomenclature) | Polyphen-2 | SIFT | Mutation Taster | Human splicing finder (v. 3.0) interpretation |
|---|---|---|---|---|---|
| L214V | c.640C>G | Probably damaging | Damaging | Disease causing | Possible creation of new donor site in exon 7 |
| A238P | c.844G>C | Probably damaging | Damaging | Disease causing | Possible creation of a new exonic ESS site in exon 7 |
| E379V | c.1136A>T | Probably damaging | Damaging | Disease causing | Possible creation of new donor site in exon 9 |
| L578I | c.1732C>A | Probably damaging | Damaging | Disease causing | Possible creation of a new exonic ESS site in exon 13 |
| F587L | c.1762T>G | Probably damaging | Damaging | Disease causing | Possible creation of a new exonic ESS site in exon 13 |
| L926W | c.2777T>G | Probably damaging | Damaging | Disease causing | Could affect splicing due to broken ESE site in exon 17 |
| R1325K | c.3974G>A | Benign | Tolerated | Polymorphism | Could affect splicing due to broken ESE site in exon 25 |
| R1453Q | c.4358G>A | Benign | Tolerated | Disease causing | Possible creation of new acceptor site in exon 27 |
The PolyPhen-2 score ranges from 0.0 (tolerated) to 1.0 (deleterious). Variants with scores of 0.0 are predicted to be benign. Values closer to 1.0 are more confidently predicted to be deleterious. PolyPhen-2 and SIFT scores use the same range, 0.0 to 1.0, but with opposite meanings. A variant with a SIFT score of 1.0 is predicted to be benign. ESS, exonic splicing enhancer; ESE, exonic splicing silencer; HGVS, Human Genome Variation Society.
Risk of cystic fibrosis or CFTR-related disorders (CFTR-RD) to the offspring: Of the cohort, 53 men and 13 females who harboured a mild or a severe variant were at risk of transmitting at least one mutant variant to the offspring. Among them, nine couples were both carriers of the CF variant (Supplementary Table V). Three CBAVD men had compound heterozygous CFTR gene mutations L926W/c.1210-12[5] and c.1521_1523delCTT (F508del)/L578I, and their female partner harboured c.1210-12[5] or a CFTR gene mutation, increasing the risk by 50 per cent for the offspring who inherit two CFTR gene variants. Six CBAVD men had one CFTR gene variant and their female partners harboured c.1210-12[5], thereby increasing the risk by 25 per cent for the offspring to inherit two CFTR gene variants.
| Couple number | CBAVD genotype | Female genotype |
|---|---|---|
| 1 | c.[1210-34TG[12];1210-12T[5]] | c.[1210-34TG[12];1210-12T[5]] |
| 2 | c.[1210-34TG[12];1210-12T[5]]/L926W | c.[1210-34TG[12];1210-12T[5]] |
| 3 | R709X/- | c.[1210-34TG[12];1210-12T[5]] |
| 4 | c.[1210-34TG[12];1210-12T[5]] | c.[1210-34TG[12];1210-12T[5]] |
| 5 | c.[1210-34TG[12];1210-12T[5]]/L926W | c.[1210-34TG[13];1210-12T[5]] |
| 6 | R1453Q/- | c.[1210-34TG[13];1210-12T[5]] |
| 7 | F508del/L578I | A1285V/- |
| 8 | R668C/- | c.[1210-34TG[12];1210-12T[5]] |
| 9 | F508del/- | c.[1210-34TG[12];1210-12T[5]] |
Outcome of intracytoplasmic sperm injection (ICSI): Among the cohort of CBAVD men who were mild or severe variant (n=53), 21 underwent at least one cycle of ICSI (40%). Among these couples, four successful pregnancies with live births including one twin pregnancy were recorded. One couple opted for adoption after ICSI failures. In nine couples wherein both partners were carriers of CFTR mutations/variants, six underwent ICSI, with two successful pregnancies resulting in live birth.
Discussion
In the present study, a heterogeneous spectrum of CFTR gene variants is reported with 66.3 per cent frequency in Indian men with CBAVD, which is lower than that in the Caucasian population1. CBAVD men were classified as patients with: (i) one variant (51.25%), (ii) two variants (15%) and (iii) no mutations (33.7%). Two CFTR gene mutations detected in 15 per cent of the CBAVD men, were comparable to those found in other studies131920. The most common mutation, F508del, was observed at a lower allelic frequency (8.75%) as compared to that of the Caucasian population1 but was higher than that of Chinese2, Japanese21 and Taiwanese22 populations and was comparable with that of north Indian CBAVD men458. On the contrary, IVS-9 c.1210-12[5] variant was reported at a higher frequency (42.5%) compared to that of studies in literature459. Failure to detect CFTR gene variants could be either due to the presence of large rearrangements such as exon deletion, insertion and duplications2324 or because of genetic aetiology other than CFTR gene. Recent studies suggest that gene ADGRG2 at a frequency of 11-15 per cent in CBAVD cases is negative for CFTR gene mutations25.
Out of the ten novel sequence variants, eight were located in highly conserved regions, which could functionally alter CFTR protein organization. Two novel splice-site variants (c.1-30C>G and c.317+2T>G) were predicted to induce splicing error and thus damage the translated CFTR protein. R75Q mutation detected in 4.2 per cent is a known variant for chronic obstructive pulmonary disease (COPD)26. R709X detected in 3 per cent of CBAVD men is predicted to result in a truncated and non-functional CFTR protein11. In addition, in vitro functional assays are warranted to provide significant insight regarding the pathogenicity of the identified variants. Although CBAVD men can achieve biological paternity via ICSI, they are at a higher risk of transmitting lethal mutations to offspring727. A few studies have attempted to screen for CF female carrier status in the Indian population. Being said that, due to the increasing burden of mutations in the CFTR gene, it has becomes difficult to provide precise molecular diagnosis and accurate genetic counselling to CBAVD patients. Considering the functional significance of the identified known and novel CFTR gene variants, genetic counselling was provided by assessing female CF carrier status. In the present study a heterozygous A1285V variant was identified in one female whose male partner had a heterozygous F508del mutation. Interestingly, A1285V variant has been reported as a novel mutation in Indian CBAVD male9. Since both the partners were carriers of CFTR gene mutation, there was a 25 per cent risk of having a child with classic CF or CFTR-RD. In addition to this, eight females who were carriers of heterozygous c.1210-12[5] variant had their male partners with a mild or severe CFTR gene variant. Therefore, genetic counselling for couples in which the male partner has CBAVD is important to estimate the risks and possible genotype–phenotype correlations prior to undergoing ICSI.
In conclusion, we found the spectrum and frequency of CFTR gene variants in the Indian population to be different from that of Caucasians. Detection of female partners as CF carriers may be useful for providing genetic counselling to the couples before enrolling them for ICSI. Complete CFTR gene screening is expensive and therefore a population-specific panel should be designed based on the known and novel variants identified in the Indian population. Further studies are underway to identify the genetic basis of CFTR-negative CBAVD subgroup.
Supplementary Fig. 1
Supplementary Fig. 1 Multiple alignments (https://www.ebi.ac.uk/Tools/msa/clustalo/) of conserved CFTR amino acid sequences from multiple species (human, mouse, rabbit, bovine, sheep, monkey, pig and horse) and eight novel CFTR gene variants (L214V, A238P, E379V, L578I, F587L, L926W, R1325K and R1453Q) identified in Indian congenital bilateral absence of vas deferens males. The conserved amino acids are highlighted in red across different species.Supplementary Fig. 2
Supplementary Fig. 2 Geographic distribution of CFTR gene variants in congenital bilateral absence of vas deferens men in India identified in this study. The red arrow indicates the identified CFTR variants from the region. * indicates novel CFTR variants.Acknowledgment
The authors acknowledge Drs Vrinda V. Khole (late); Smita Mahale, Director, ICMR-NIRRH and S.D. Kholkute, Former Director, ICMR-NIRRH, Mumbai for their kind support in implementation of the study. Drs Jyotsna Gokral, Pervin Meherji (late), Anurupa Maitra are acknowledged for scientific inputs during conceptualisation of study. Dr Srabani Mukherjee, Ms Nanada Joshi, Mr C. Saravanan, Dr Parag Tamhankar, Ms Shweta, Dr Suparna Chatterjee are sincerely acknowledged for technical assistance with Sanger sequencing. Mr Ashok Vadigopula is acknowledged for assistance in semen analysis and blood collection. Dr A R Pasi is acknowledged for assistance in statistical analysis and Dr Nobhojit Roy is acknowledged for assistance in coordinating with funding agency, Board of Research in Nuclear Sciences, Department of Atomic Energy, Government of India
Financial support & sponsorship: This study was partially funded by the intramural grant of ICMR (NIRRH/RA/782/07/2019) and extramural grant of Board of Research in Nuclear Sciences (2013/37B/12/BRNS), Department of Atomic Energy, Government of India.
Conflicts of Interest: None.
References
- Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros. 2011;10(Suppl 2):S86-102.
- [Google Scholar]
- Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) in Chinese patients with congenital bilateral absence of vas deferens. J Cyst Fibros. 2012;11:316-23.
- [Google Scholar]
- CFTR mutations in men with congenital bilateral absence of the vas deferens (CBAVD): A systemic review and meta-analysis. Hum Reprod. 2012;27:25-35.
- [Google Scholar]
- Heterogenous spectrum of CFTR gene mutations in Indian patients with congenital absence of vas deferens. Hum Reprod. 2009;24:1229-36.
- [Google Scholar]
- Heterogeneous spectrum of mutations in CFTR gene from Indian patients with congenital absence of the vas deferens and their association with cystic fibrosis genetic modifiers. Mol Hum Reprod. 2014;20:827-35.
- [Google Scholar]
- Cystic fibrosis transmembrane conductance regulator (CFTR) gene abnormalities in Indian males with congenital bilateral absence of vas deferens & renal anomalies. Indian J Med Res. 2016;143:616-23.
- [Google Scholar]
- The CFTR gene mild variants poly-T, TG repeats and M470V detection in Indian men with congenital bilateral absence of vas deferens. Andrologia. 2018;50:e12858.
- [Google Scholar]
- Cystic fibrosis, CFTR gene, and male infertility. In: Singh R, Singh K, eds. Male infertility: understanding, causes and treatment. Singapore: Springer Singapore; 2017. p. :131-50.
- [Google Scholar]
- Mutation studies in the CFTR gene in Asian Indian subjects with congenital bilateral absence of vas deferens: Report of two novel mutations and four novel variants. Genet Test Mol Biomarkers. 2011;15:307-12.
- [Google Scholar]
- Molecular analysis of the IVS8-T splice variant 5T and M470V exon 10 missense polymorphism in Iranian males with congenital bilateral absence of the vas deferens. Mol Hum Reprod. 2006;12:469-73.
- [Google Scholar]
- Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med. 1995;332:1475-80.
- [Google Scholar]
- Heterogeneity for mutations in the CFTR gene and clinical correlations in patients with congenital absence of the vas deferens. Hum Reprod. 2000;15:1476-83.
- [Google Scholar]
- Characterization of cystic fibrosis conductance transmembrane regulator gene mutations and IVS8 poly(T) variants in Portuguese patients with congenital absence of the vas deferens. Hum Reprod. 2004;19:2502-8.
- [Google Scholar]
- Mutations of the CFTR gene in Turkish patients with congenital bilateral absence of the vas deferens. Hum Reprod. 2004;19:1094-100.
- [Google Scholar]
- Congenital absence of the vas deferens. The fertilizing capacity of human epididymal sperm. N Engl J Med. 1990;323:1788-92.
- [Google Scholar]
- Congenital bilateral absence of the vas deferens as an atypical form of cystic fibrosis: Reproductive implications and genetic counseling. Andrology. 2018;6:127-35.
- [Google Scholar]
- Proportion of cystic fibrosis gene mutations not detected by routine testing in men with obstructive azoospermia. JAMA. 1999;281:2217-24.
- [Google Scholar]
- WHO laboratory manual for the examination and processing of human semen (5th ed). Geneva: WHO; 2010. p. :14.
- Heterogeneity of reproductive tract abnormalities in men with absence of the vas deferens: Role of cystic fibrosis transmembrane conductance regulator gene mutations. Fertil Steril. 1998;70:724-8.
- [Google Scholar]
- The genetic basis of congenital bilateral absence of the vas deferens and cystic fibrosis. J Androl. 1994;15:1-8.
- [Google Scholar]
- CFTR gene mutations in Japanese individuals with congenital bilateral absence of the vas deferens. J Cyst Fibros. 2003;2:14-8.
- [Google Scholar]
- Cystic fibrosis transmembrane conductance regulator gene screening and clinical correlation in Taiwanese males with congenital bilateral absence of the vas deferens. Hum Reprod. 2004;19:250-3.
- [Google Scholar]
- Large genomic rearrangements in the CFTR gene contribute to CBAVD. BMC Med Genet. 2007;8:22.
- [Google Scholar]
- Detection of cystic fibrosis transmembrane conductance regulator (CFTR) gene rearrangements enriches the mutation spectrum in congenital bilateral absence of the vas deferens and impacts on genetic counselling. Hum Reprod. 2007;22:1285-91.
- [Google Scholar]
- Truncating mutations in the adhesion G protein-coupled receptor G2 gene ADGRG2 cause an X-linked congenital bilateral absence of vas deferens. Am J Hum Genet. 2016;99:437-42.
- [Google Scholar]
- High frequency of the R75Q CFTR variation in patients with chronic obstructive pulmonary disease. J Cyst Fibros. 2004;3:189-91.
- [Google Scholar]
- Molecular basis of cystic fibrosis disease: An Indian perspective. Indian J Clin Biochem. 2010;25:335-41.
- [Google Scholar]




