Translate this page into:
Abnormal haemoglobins: detection & characterization
Reprint requests: Dr Henri Wajcman, INSERM, Unité U955, Créteil, France e-mail: Henri.Wajcman@inserm.fr
-
Received: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Haemoglobin (Hb) abnormalities though quite frequent, are generally detected in populations during surveys and programmes run for prevention of Hb disorders. Several methods are now available for detection of Hb abnormalities. In this review, the following are discussed: (i) the methods used for characterization of haemoglobin disorders; (ii) the problems linked to diagnosis of thalassaemic trait; (iii) the strategy for detection of common Hb variants; and (iv) the difficulties in identification of rare variants. The differences between developing and industrialized countries for the strategies employed in the diagnosis of abnormal haemoglobins are considered. We mention the limits and pitfalls for each approach and the necessity to characterize the abnormalities using at least two different methods. The recommended strategy is to use a combination of cation-exchange high performance chromatography (CE-HPLC), capillary electrophoresis (CE) and when possible isoelectric focusing (IEF). Difficult cases may demand further investigations requiring specialized protein and/or molecular biology techniques.
Keywords
Haemoglobin variant
neonatal screening
RBC density
sickle cell
thalassaemia

Introduction
Haemoglobin (Hb) abnormalities are the most frequent genetic disease, affecting approximately 7 per cent of the world population1. Today, abnormal Hbs are generally discovered during a systematic study performed within programmes for prevention of thalassaemias or sickle cell disease. In several regions (India, Turkey, Irak, Iran, Gaza strip, Saudi Arabia, Cyprus, etc.), these are found during a premarital screening. In other regions, like west Europeans countries, the research for the main Hb disorders is often limited to populations at risk. It is done either as preconceptional or neonatal screening programmes.
In all cases, besides the study of the Hb, a first essential test that need to be performed is a complete blood cell count (CBC) looking mostly for anaemia, microcytosis and, hypochromia. If necessary, it should be completed by a control of the blood iron status. Several high throughput methods, now available, such as cation-exchange high performance chromatography (CE-HPLC)23 or more recently capillary electrophoresis (CE)4 are generally employed. Nevertheless, electrophoretic studies [cellulose-acetate electrophoresis or isoelectric focusing (IEF)] are still performed in many laboratories5. In some developing countries, the Naked Eye Single Tube Red Cell Osmotic Fragility Test (NESTROFT), a cost effective, rapid and reliable screening test for detection of β-thalassaemia trait, is largely used as a first approach6.
The investigation of a patient presenting with a haematological disorder such as a chronic haemolytic anaemia, an unexplained polycythaemia or a permanent cyanosis is another, completely different condition, leading to the discovery of an abnormal Hb.
In this review, we will discuss (i) the methods used for characterization of haemoglobin disorders; (ii) the problems linked to diagnosis of thalassaemic trait; (iii) the strategy for detection of common Hb variants; and (iv) the difficulties in identification of rare variants.
Methodological aspects: The main tests used for phenotype characterization
Electrophoresis, a test based on the migration of electrically charged molecules under an applied electric field, occupies one of the most important places in the history of abnormal Hb detection. Hb S [β6 Glu>Val], the first abnormal Hb described, was discovered in 1949 by Pauling et al using moving boundary electrophoresis7. Zone electrophoresis performed on cellulose acetate strips (CAE) is still used in many clinical laboratories. This technique has a resolution lower than that of IEF, but because of its simplicity remains among the more popular methods used in Hb screening. In this technique, the Hb molecules are separated at alkaline pH. Under these conditions all Hbs have a negative charge and migrate towards the cathode. Hb S, which has an additional positive charge compared to Hb A migrates more slowly5.
In IEF, a pH gradient is established by carrier ampholytes submitted to an electric current and the Hb molecules migrate across this gradient until they reach the position where their net charge is zero (isoelectric point)8. The molecules will then concentrate in a sharp band as illustrated in Fig. 1. This technique allows separating molecules with isoelectric points differing only by 0.02 pH unit. Hb D-Punjab [β121 Glu>Gln], not detectable from Hb S by CAE, could easily be recognized by this technique. The disadvantage of this method is its relatively high cost and the requirement for a well-experienced laboratory staff.

- Isoelectric focusing analysis of red blood cells lysates containing different Hb variants, technical details are available in Ref. 5. Line 1: β thalassaemia trait; Line 2: Normal adult; Line 3: Homozygous β+ thal; Line 4: HbA/Hb Korle Bu; Line 5: HbA/HbD-Punjab; Line 6: HbA/HbO-Arab; Line 7: Normal newborn; Line 8: HbA/HbE; Line 9: HbA/HbC; Line 10: HbA/HbS; Line 11: HbS/HbC.
Electrophoresis of Hbs on agar at acidic pH has been introduced some forty years ago9. It is not primarily sensitive to the charge of the mutated residue but to structural modifications of positively charged regions of the Hb molecule interacting with the agaropectin contained in the gel10. This property is of special interest to distinguish Hb S from other variants displaying mobility close to that of Hb S on CAE or IEF.
Capillary electrophoresis has been recently adapted to Hb study11. This technique shares several advantages with CE-HPLC: it is a high resolution method associated with semi-automation, on-line detection and direct quantification of normal and abnormal Hb fractions. The Capillarys® system (Sebia, Evry, France) is equipped with eight capillaries in parallel, allowing multiple and simultaneous sample analysis. Each capillary can be used at least 3000 times12.
Cation-exchange HPLC is considered as the method of choice to quantify the various normal and abnormal Hb fractions13. This is the method of reference for measuring Hb A1C for monitoring of diabetes mellitus. It is also of general use for measurement of the levels of Hb A2, Hb F and of several abnormal Hbs23. Automated apparatus have been developed since several years ago for large series measurement. Cation-exchange HPLC and CE are the two methods recommended for detection of thalassaemic traits necessary for genetic counselling since these are the ones allowing a precise quantification of Hb A2, Hb F and abnormal Hb fractions. To make the diagnosis of a thalassaemic trait or quantify the abnormal Hb fraction, the use of densitometry scanning of electrophoretic pattern is a too imprecise method to be used today.
Three methods are proposed for neonatal screening of sickle cell disease (Fig. 2). The first one is an adaptation of IEF which allows the simultaneous study of about 90 samples on a single plate, using a central cathode and two anodes14. At a glance, any band with abnormal mobility is detected as well as the absence of the normal Hb A band. This method is considered as a first line test and requires a confirmation by a second one, usually CE-HPLC. Cation-exchange-HPLC equipment devoted to neonatal screening have been developed which, in a 2.5 min programme per sample, allow quantitative detection of Hb S and of the most common Hb variants15. This approach by CE-HPLC is also suitable for neonatal diagnosis of thalassaemic patients16. Capillary electrophoresis is the third method that may be successfully used for neonatal screening17. Characterization of rare Hb variants responsible for a haematological disorder requires a completely different approach. The presence of a variant is usually obtained by IEF, which shows either a band with a clearly different isoelectric point or an abnormal aspect of the Hb A band suggesting that it contains two fractions (Fig. 1). Cation-exchange HPLC or CE may confirm the presence of this abnormal component or reveal an abnormal Hb, not detected by IEF. The use of additional methods is frequently required. For example, reversed-phase high-performance liquid chromatography (RP-HPLC) reveals differences in hydrophobicity allowing discrimination between variants displaying identical charges. This approach also indicates which globin chain is affected18.
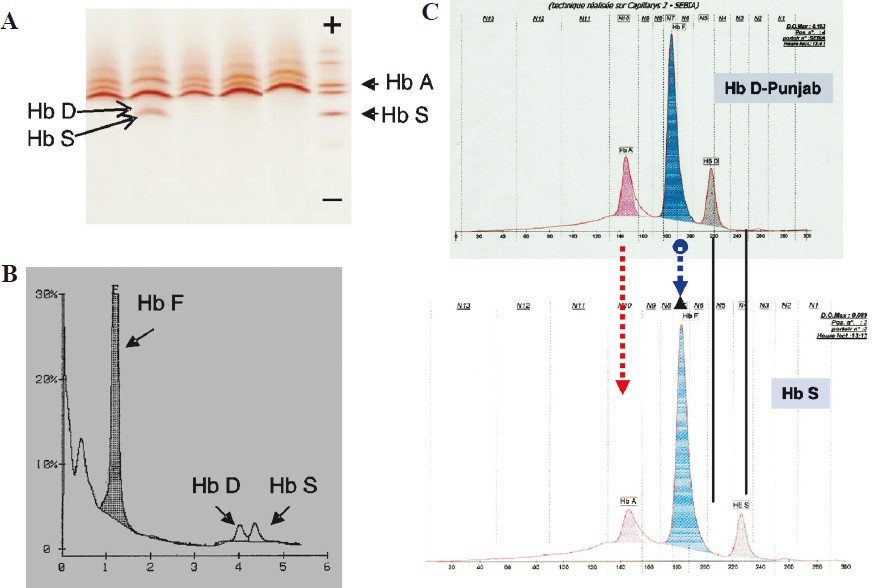
- Neonatal diagnosis for Hb S and Hb D-Punjab. Analysis of RBC lysates obtained from a dried blood spot obtained during the neonatal screening programme done in our laboratory. Analysis by two methods is required for the diagnosis. (A) Isoelectric focusing: Hb S and Hb D-Punjab have close isoelectric points and the resolution between the two bands is poor. The absence of HbA and thickening of HbS band help to diagnose. (B) Cation-exchange HPLC shows two abnormal peaks: one is eluted in S window and the other in the D window. (C) Capillary electrophoresis: (above): newborn heterozygous for Hb D-Punjab and (below): newborn heterozygous for Hb S. The two Hbs have different migrations.
In well-equipped laboratories of industrialized countries mass spectrometry methods may be used to determine the structural abnormalities. Electrospray mass spectrometry may reveal a globin chain with an abnormal mass and, from the mass difference observed, the type of amino acid exchanged could often be deduced. Methods such as MALDI-TOF or mass-mass spectrometry in which the globin chains are cleaved in small fragments will show a single fragment, or a family of fragments from which the exact substitution could be determined1920. Today, the facility for doing a DNA sequence analysis and its relatively low cost changed completely the approach for characterization of some rare Hb variants associated with a severe pathological phenotype not detected by conventional methods.
There are many different PCR-based techniques that can be used to diagnose the globin gene mutations, including dot-blot analysis, reverse dot-blot analysis, the amplification refractory mutation system (ARMS), high resolution melting (HRM), gap-PCR and restriction endonuclease analysis2122. Microarray-based human globin gene mutation detection methods have been recently reviewed23.
Problems linked to diagnosis of β-thalassaemia trait
The key element in the diagnosis of a β-thalassaemia trait is the presence of non-iron deficient microcytic hypochromia anaemia. The screening of thalassaemia trait in the areas with limited laboratory facilities is often done by NESTROFT test6. Despite its sensitivity and rapidity, in around one out of four cases of iron deficiency anaemia, this test leads to a false positive result24. The best approach to screen the thalassaemia traits is, therefore, determining the Hb A2 level. The large majority of β-thalassaemia carriers present with a high Hb A2 level, and this often accompanied with a moderate increase in Hb F level25.
Hb A2 level is correctly measured by CE-HPLC but it is necessary to keep in mind that the normal value depends upon the experimental procedures used26. From one chromatographic system to another slight differences may be observed and values within the normal range in a given system may well fall out of the normal range in another system. Thus the normal values of the laboratory where the test is performed should always be specified.
The Hb A2 level may be modified by many factors. The most frequent problem is the co-existence of an iron deficiency which may even normalize the Hb A2 level requiring a novel Hb assay after iron deficiency treatment. In β-thalassaemia carriers presenting with a normal Hb A2 level, the most frequent cause is a co-inherited δ-globin abnormality. The thalassaemic δ alleles are not exceptional affecting at least more than one per cent of general population27. The most frequent δ thalassaemic variant is Hb Yialoussa [δ27 Ala>Ser] which is quite frequent in Mediterranean region28. Other δ- and α-globin chain abnormalities lead to different type of Hb A2. In these cases one should add the two Hb A2 fractions to obtain the total Hb A2 level. In some cases the abnormal Hb A2 fraction may only be visualized by using another analytical test. Conversely, falsely increased levels of Hb A2 may result from the co-existence of a variant with electrophoretic or chromatographic properties close to that of Hb A2. As a rule, this situation has to be verified when a level of Hb A2 higher than 8 per cent is observed. The presence of a thalassaemia will generally be ruled out by considering haematological data showing absence of microcytosis and hypochromia. Nevertheless, two common variants, which elute on CE-HPLC in the Hb A2 window, are really thalassaemic Hbs. The first one is Hb E, frequent in populations from South East Asia, and the second is Hb Lepore. Both variants are easy to distinguish by their electrophoretic behaviour. Since many other thalassaemic defects are present in the same populations as Hb E, a large variety of clinical phenotype associated with Hb E is observed29. These range from very mild ones, the simple heterozygous state for Hb E, to severe ones, which associate Hb E to β° thalassaemia.
Another important element for the diagnosis of a β-thalassaemia trait is the presence of Hb F. In the past, the classical way to determine its percentage was to perform a resistance to alkali denaturation test. As other kinetic techniques, it needs to be performed under rigorous experimental conditions with a precise respect of the timing of the reaction30. This method measures together Hb F and its acetylated fraction, which amounts to about 10 per cent of it. Now CE-HPLC and CE allow a direct measurement of the Hb F fractions. Depending on the system used, some other Hb adducts may elute together with Hb F and increase slightly the area of the elution peak. In these systems acetylated Hb F elutes generally in another position than Hb F and is therefore, not included in the result. Thus, in the same sample the percentage of Hb F will differ according to the method used. This is not really important since in an adult any value of Hb F higher than 1 per cent should be considered as unusual. Increased Hb F level may indicate a β-thalassaemia trait but it could also be the result of a hereditary persistence of foetal Hb (HPFH) or of another condition, such as diabetes mellitus, or pregnancy.
Diagnosis of an α thalassaemia will be suspected by haematological indices showing some microcytosis and hypochromia, with a degree depending upon the type of thalassaemia. The Hb A2 percentage is theoretically decreased in proportion with the number of defective α genes. Abnormal Hb tetramers are observed in the more severe forms: Hb Bart's (γ4) in newborns, and Hb H (β4) in adults. The presence of an α thal-2 in which only one of the four α genes is inefficient is one of the most common genetic modifications observed worldwide. An interaction of this situation with other globin abnormalities should always be considered as a possible modulator factor31. In a carrier of a β- thalassaemic trait the co-existence of an α-thalassaemia will partially compensate the disequilibrium in globin synthesis; due to a lesser amount of toxic free α globin chain bound to the red cell membrane, the hypochromic anaemia will be much better tolerated. Conversely the association of a β-thalassaemic trait with additional α genes may lead to a β-thalassaemia intermedia phenotype32.
Problems linked to diagnosis of common Hb variants
Hb S is, worldwide, the most frequent, clinically severe Hb variant. It is specially encountered in populations from African ancestry, essentially carried on chromosomes with Senegal, Benin or Bantou haplotypes33. In India and Arabic countries, Hb S has frequently another origin, since in many cases it is found in a haplotype named Arabo-Indian and its characteristic is to be linked to a relatively high Hb F level. In these regions, the Hb S allele being much less frequent than in Africa, the patients who appear as homozygous are frequently compound heterozygous for Hb S and a β° thalassaemia. The βS allele in homozygous status is mostly found in some tribal populations of India34.
Historically the diagnosis of Hb S was performed by electrophoretic methods. If IEF is still used in many laboratories, it becomes the rule to be accomplished by CE-HPLC or CE. Since Hb S shares its electrophoretic and chromatographic properties with several variants it is necessary, in the absence of familial history of Hb S, to do a confirmatory test. In the past, the solubility test was considered as the golden standard since Hb S is the only abnormal Hb, which is insoluble at high concentrations when deoxygenated. This test is now commercially difficult to get, and the large majority of the clinical laboratories do not want to prepare their own reagents. Thus combination of several methods needs to be used to confirm the diagnosis. A good compromise is to perform CE-HPLC and CE, or acid agar electrophoresis.
Only in some exceptional cases in whom another mutation is associated in cis with that of Hb S the heterozygous carriers can also present a severe pathologic phenotype. Examples are provided by the heterozygous carriers of Hb S-Antilles [β6 Glu>Val and β23Val>Ile]35 or Hb S-Oman [β6Glu>Val and β121Glu>Lys]36 in whom the second mutation enhances sickling. Recently, an unstable β allele (βS-San Martin) associating in cis the βS allele to a mutation causing instability of the haemoglobin was described37.
A few common variants, which lead to sickle cell anaemia when associated to Hb S have to be imperatively characterized, e.g. Hb D-Punjab and O-Arab. Both affect position β121 (GH4) where the Glu is replaced by a Gln in the first case, and by a Lys in the second one. This residue is located at the outside of the Hb tetramer in a region, which participates in the building of the Hb S fiber during sickling. These two mutations facilitate polymerization and, as a result, compound heterozygotes may be, clinically, as severe as homozygous Hb S. Hb D-Punjab cannot be easily recognized from Hb S by using conventional CAE. For this method the same is true for Hb O-Arab that may be confused with Hb E or Hb C. In contrast, CE-HPLC or CE allows an easy identification of these two variants. This consideration is especially important for neonatal screening (Fig. 2). Since Hb D-Punjab is the only variant with Hb D mobility, which leads to a severe disease when associated with HbS, its identity must be recognized. Other variants such as Hb D-Iran, Hb Korle-Bu are completely harmless. For these variants, typical CE-HPLC profiles are observed. Fig. 3 shows CE-HPLC elution profiles of carriers of Hb O-Arab in various different compound heterozygous states. In the heterozygous or homozygous forms, Hb D-Punjab or O-Arab have no clinical consequences but these need to be identified in programmes for prevention of sickle cell anaemia. The individuals compound heterozygous for one of these abnormalities and a β-thalassaemia allele are haematologically identical to a simple β-thalassaemia carrier.
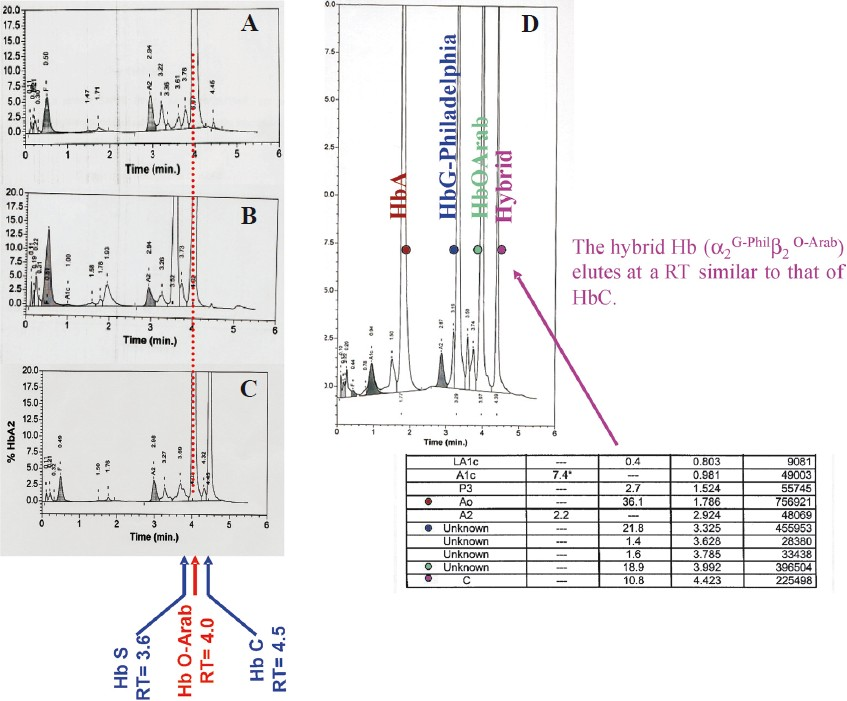
- CE-HPLC: Analysis of haemolysates from patients presenting with different associations involving Hb O-Arab. (A) Compound heterozygous Hb O-Arab/β°thalassaemia. (B) Compound heterozygous Hb S/HbO-Arab. (C) Compound heterozygous Hb C/Hb O-Arab. (D) Heterozygote Hb O-Arab associated with G-Philadelphia. The hybrid Hb (α2G-Philβ2O-Arab) elutes in C window.
Presence of Hb S in homozygous or compound heterozygous with Hb variants, involved in sickling, modifies the RBC density distribution which can be studied easily using phthalate density gradient3839. Fig. 4 illustrates the different profiles that could explain the clinical heterogeneity of patients presenting with sickle cell anaemia associated with different genotypes. It has to be pointed out that this simple and low cost technique may be helpful to diagnose different α or β-thalassaemia associated to other Hb variants.
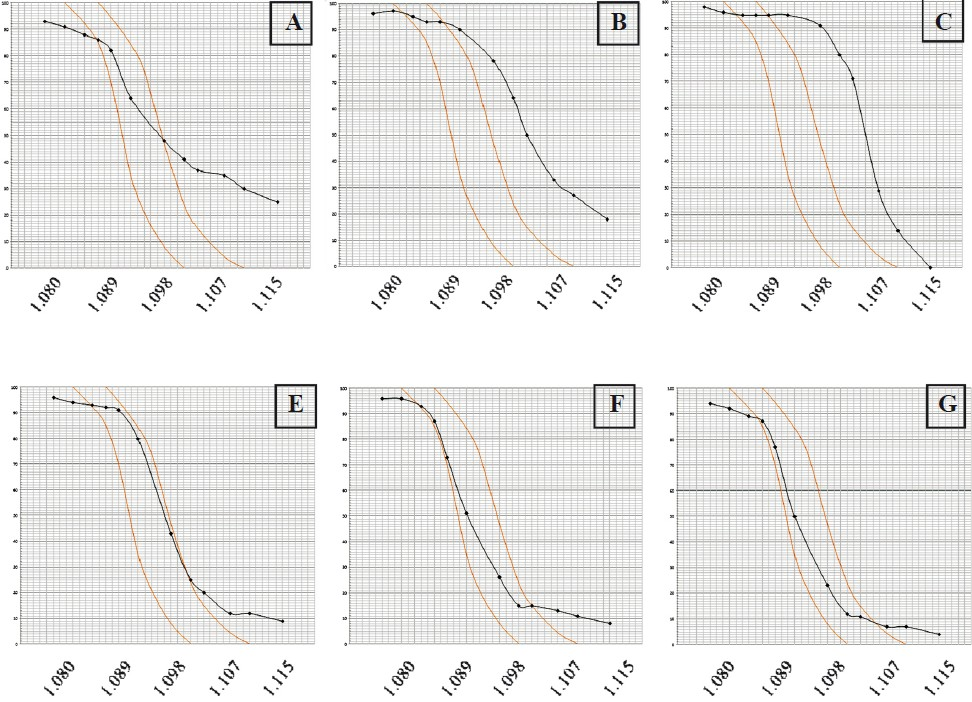
- Red blood cell density profiles of patients with Hb S associated to various genotypes (technical details are available in Ref. 38). The percentage of cells denser than the phthalate index (y axis) is plotted against the density of the phthalate mixtures (x axis) used expressed in g/ml. Limits of normal distribution are shown in each graph. (A) Homozygous Hb S. Dense cells and young cells (reticulocytes) are observed at the right and left side of the distribution, respectively. (B) Compound heterozygous Hb S/Hb O-Arab. The high percentage of dense cells is observed explaining the severity of this association. (C) Compound heterozygous Hb S/Hb C. (D) Homozygous Hb S associated to an α thal-2. (E) Homozygous Hb S associated to an α thal-1. (F) Compound heterozygote Hb S/β°-thalassaemia. The Curve is shifted to the left when Hb S is associated to a thalassaemic allele.
Hb C and Hb E are also two frequent variants. The first one, which originates from the region of Upper Volta is frequent in Africa, while the second one is common in South East Asia40. Since, both result from the replacement of a glutamic acid by a lysine these variants share some electrophoretical properties and it is difficult to distinguish these by CAE. The diagnosis between Hb C and Hb E can be established by CE-HPLC and CE, which show a different profile for these two variants. In Thailand several cases, initially considered as Hb E, were characterized finally as Hb C using above mentioned techniques41. Hb C and Hb E both lead to some degree of microcytosis. As a consequence of low expression of βE allele, Hb E is associated with hypochromia, whereas HbC with hyperchromia because of some dehydration of the red blood cell. Regarding the high frequency of Hb S and Hb C in the population originated from Africa, compound heterozygotes are not rare. And due to increasing intracellular concentration of total Hb (high MCHC linked to Hb C) the compound heterozygotes Hb S/Hb C sickle. Considering their low MCHC, the rare cases of Hb S/Hb E have a mild degree of clinical symptoms. The distribution of RBCs of heterozygous individuals of Hb E and Hb C can be also easily distinguished using phthalate density gradient.
Problems linked to diagnosis of rare Hb variants
Among the rare Hb variants some may have a high local incidence as Hb C, Hb S, Hb E or the various thalassaemic defects because these bring some protective effect against malaria. This is likely the case of these unstable α chain variants which display in the heterozygous state some mild α thalassaemic features. Among those the most frequent is perhaps Hb Constant Spring resulting from a mutation of the termination codon, leading to a low expressed elongated unstable α chain42. Many variants belonging to this group are now known and some have been reported in Asian populations43.
Among the other rare variants several groups may be individualized. The first group is made by those variants which are without any clinical effects and are fortuitously found during screening programmes. The only reason that leads to characterize these is to eliminate a problem of possible interaction with Hb S or with a thalassaemic syndrome. These will be found because of a different electrophoretic or chromatographic behaviour. By comparing their properties in a battery of tests (IEF, CE-HPLC, RP-HPLC, globin chain electrophoresis, electrospray mass measurement) it is often possible to identify these without need for a complete protein sequence analysis or DNA sequencing.
The second group is that of rare Hb variants causing haematological disorders. Even when present in the heterozygous state, these lead to dominant manifestations. A dominant phenotype of haemolytic anaemia is always observed in the case of unstable Hbs. Historically, the concept of this group of Hbs arose some 50 years ago from the study of patients suffering from chronic non-spherocytic haemolytic anaemia. Their haemolysate displayed a normal electrophoretic picture, but when incubated at 50°C revealed the presence of a Hb component, which precipitated faster than Hb A44. Blood smears, either spontaneously or after incubation with an oxidant dye, showed inclusion bodies, made from Hb precipitates. Later on it was demonstrated that these Hb variants displayed structural modifications in some specific regions of the Hb molecule such as the haem pocket or the α1β1 interface. Other variants having together electrophoretic abnormalities and instability were found later on45. From a biological and clinical point of view, unstable Hbs form a very heterogeneous group. Each of the mutations responsible for an unstable Hb affects only a limited number of individuals in a few families, usually unrelated. De novo mutations are frequently observed since these mutations are always a cause of disease. These do not bring any selective advantage and are, thus, eliminated in the forthcoming generations. In industrialized countries the founder effects are usually identified since any patient suffering from haemolytic anaemia will be investigated until the exact aetiology of the disease will be explained.
Conclusions
Today, detection of Hb mutants is usually easily performed by automatic methods of protein study (CE-HPLC and CE). This led to the discovery of an impressive list of variants which are reported on the Hbvar database (http://globin.cse.psu.edu/hbvar/menu.html)45. The majority of Hb variants fortuitously discovered are of minimal clinical interest. Conversely, those found during the course of a haematological disorder bring frequently the aetiological answer for the disease. Often unusual clinical presentations may be explained by the interaction of several Hb abnormalities and their identification may require further investigations.
Authors thank Claude Prehu, Mireille Mathis, Jean Riou, Christian Godart and Didier Hurtrel of Hemoglobinopathies Laboratory of Henri Mondor University hospital for their contribution to this work by providing some helpful data, and acknowledge Josiane Bardakdjian and Michel Bahuau for providing the data in neonatal screening.
References
- Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79:704-12.
- [Google Scholar]
- Cation-exchange HPLC evaluated for presumptive identification of haemoglobin variants. Clin Chem. 1997;43:34-9.
- [Google Scholar]
- HPLC retention time as a diagnostic tool for haemoglobin variants and haemoglobinopathies: a study of 60000 samples in a clinical diagnostic laboratory. Clin Chem. 2004;50:1736-47.
- [Google Scholar]
- Evaluation of a new Sebia kit for analysis of haemoglobin fractions and variants on the Capillary system. Clin Chem Lab Med. 2006;44:340-5.
- [Google Scholar]
- Electrophoretic Methods for Study of Haemoglobins. In: Nagel RL, ed. Methods in molecular medicine. Vol 82. Totowa, NJ: Humana Press Inc; 2003. p. :93-100. Haemoglobin disorders: Molecular methods and protocols
- [Google Scholar]
- NESTROFF as a screening test for detection of Beta-thalassaemia trait. Indian J Pathol Microbiol. 2002;45:265-7.
- [Google Scholar]
- Isoelectric focusing: Theory, methodology and applications. Amsterdam, The Netherlands: Elsevier; 1983.
- Hemoglobin mobility in citrate agar electrophoresis: its relationship to anion binding. Hemoglobin. 1982;6:199-208.
- [Google Scholar]
- Interaction of human hemoglobin and its variants with agar. Science. 1983;221:175-8.
- [Google Scholar]
- Evaluating five dedicated automatic devices for hemoglobinopathy diagnostics in multi-ethnic populations. Int J Lab Hematol. 2009;31:484-95.
- [Google Scholar]
- Evaluation of an automated capillary electrophoresis system in the screening for hemoglobinopathies. Clin Lab. 2009;55:217-21.
- [Google Scholar]
- Strategy linking several analytical methods of neonatal screening for sickle cell disease. J Med Screen. 2001;8:8-14.
- [Google Scholar]
- Implementing neonatal screening for hemoglobinopathies in the Netherlands. J Med Screen. 2010;17:58-65.
- [Google Scholar]
- A brief review on newborn screening methods for hemoglobinopathies and preliminary results selecting beta thalassaemia carriers at birth by quantitative estimation of the HbA fraction. Clin Biochem. 2009;42:1780-5.
- [Google Scholar]
- Newborn screening for hemoglobinopathies using capillary electrophoresis technology: Testing the Capillarys® Neonat Fast Hb device. Clin Biochem. 2010;43:1345-50.
- [Google Scholar]
- Globin chain analysis: an important tool in phenotype study of hemoglobin disorders. Clin Biochem. 2009;42:1802-6.
- [Google Scholar]
- New results of hemoglobin variant structure determinations by fast atom bombardment mass spectrometry. Hemoglobin. 1990;14:529-48.
- [Google Scholar]
- Use of combined mass spectrometry methods for the characterization of a new variant of human haemoglobin: the double mutant haemoglobin villeparisis b77(EF1) His!Tyr, b80(EF4) Asn>Ser. J Am Soc Mass Spectrom. 1996;7:163-7.
- [Google Scholar]
- DNA diagnosis of hemoglobin mutations. In: Nagel RL, ed. Methods in molecular medicine. Vol 82. Totowa, NJ: Humana Press Inc; 2003. p. :101-16. Haemoglobin disorders: Molecular methods and protocols
- [Google Scholar]
- Rapid identification of HBB gene mutations by high-resolution melting analysis. Clin Biochem. 2009;42:1667-76.
- [Google Scholar]
- An overview of current microarray-based human globin gene mutation detection methods. Hemoglobin. 2007;31:289-311.
- [Google Scholar]
- Effectiveness of red cell osmotic fragility test with varying degrees of saline concentration in detecting beta-thalassaemia trait. Singapore Med J. 2008;49:823-6.
- [Google Scholar]
- Evaluation of the Bio-Rad VARIANT_ II HbA2/HbA1C Dual Program for measurement of hemoglobin concentrations and detection of variants. Clin Chem Lab Med. 2005;43:237-43.
- [Google Scholar]
- Genotype-phenotype relationship of the δ-thalassaemia and Hb A(2) variants: observation of 52 genotypes. Hemoglobin. 2010;34:407-23.
- [Google Scholar]
- The significance of the haemoglobin A(2) value in screening for haemoglobinopathies. Clin Biochem. 2009;42:1786-96.
- [Google Scholar]
- Frequency of alpha-globin gene triplications and their interaction with beta-thalassaemia mutations. Hemoglobin. 2009;33:124-31.
- [Google Scholar]
- Evidence for the multicentric origin of the sickle cell haemoglobin gene in Africa. Proc Natl Acad Sci USA. 1984;81:1771-3.
- [Google Scholar]
- Sickle cell disease among tribes of Andhra Pradesh and Orissa, India. Anthropol Anz. 2002;60:169-74.
- [Google Scholar]
- Hemoglobin S Antilles: a variant with lower solubility than hemoglobin S and producing sickle cell disease in heterozygotes. Proc Natl Acad Sci USA. 1986;83:9363-7.
- [Google Scholar]
- Hb S-San Martin: a new sickling haemoglobin with two amino acid substitutions [β6(A3)Glu→Val;β105(G7)Leu→Pro] in an Argentinean family. Hemoglobin. 2010;34:500-4.
- [Google Scholar]
- Determination of density distribution of red cell population. J Lab Clin Med. 1964;64:668-74.
- [Google Scholar]
- Red blood cell indices, cation content, and membrane cation transports. Hemoglobin. 1998;22:493-500.
- [Google Scholar]
- Molecular basis and hematological features of hemoglobin variants in Southern Thailand. Int J Hematol. 2010;92:445-50.
- [Google Scholar]
- Unstable and thalassemic alpha chain hemoglobin variants: a cause of Hb H disease and thalassemia intermedia. Hemoglobin. 2008;32:327-49.
- [Google Scholar]
- Idiopathic Heinz body anaemia: Hb-Bristol (beta67 (E11) Val to Asp) Br J Haematol. 1970;18:435-46.
- [Google Scholar]
- Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies. Nucl Acids Res. 2004;32:D537-41.
- [Google Scholar]




