
Translate this page into:
Drug repurposing for identification of potential inhibitors against SARS-CoV-2 spike receptor-binding domain: An in silico approach
For correspondence: Dr Sanghamitra Pati, ICMR-Regional Medical Research Centre, Nalco Square, Chandrasekharpur, Bhubaneswar 751 023, Odisha, India e-mail: drsanghamitra12@gmail.com
-
Received: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
The world is currently under the threat of coronavirus disease 2019 (COVID-19) infection, caused by SARS-CoV-2. The objective of the present investigation was to repurpose the drugs with potential antiviral activity against receptor-binding domain (RBD) of SARS-CoV-2 spike (S) protein among 56 commercially available drugs. Therefore, an integrative computational approach, using molecular docking, quantum chemical calculation and molecular dynamics, was performed to unzip the effective drug-target interactions between RBD and 56 commercially available drugs.
Methods:
The present in silico approach was based on information of drugs and experimentally derived crystal structure of RBD of SARS-CoV-2 S protein. Molecular docking analysis was performed for RBD against all 56 reported drugs using AutoDock 4.2 tool to screen the drugs with better potential antiviral activity which were further analysed by other computational tools for repurposing potential drug or drugs for COVID-19 therapeutics.
Results:
Drugs such as chalcone, grazoprevir, enzaplatovir, dolutegravir, daclatasvir, tideglusib, presatovir, remdesivir and simeprevir were predicted to be potentially effective antiviral drugs against RBD and could have good COVID-19 therapeutic efficacy. Simeprevir displayed the highest binding affinity and reactivity against RBD with the values of −8.52 kcal/mol (binding energy) and 9.254 kcal/mol (band energy gap) among all the 56 drugs under investigation.
Interpretation & conclusions:
In the current investigation, simeprevir was identified as the potential antiviral drug based on the in silico findings in comparison to remdesivir, favipiravir and other 53 drugs. Further, laboratory and clinical investigations are needed to be carried out which will aid in the development of quick therapeutics designed for COVID-19.
Keywords
Angiotensin-converting enzyme 2
COVID-19
in silico
receptor-binding domain
SARS-CoV-2
simeprevir

SARS-CoV-2 belongs to one of the largest RNA virus genomes, ranging in size from 27 to 32 kb. The viral genome of SARS-CoV-2 contains non-structural protein genes with open-reading frame (ORF) 1a, ORF1b and four key structural proteins that are encoded by spike (S), envelope (E), membrane (M) and nucleocapsid (N) genes1. Among all the structural proteins, CoV spike (S) glycoprotein has been reported to play the most crucial role in viral attachment, fusion and entry and also used as a key target for the production of antibodies, vaccines and entry inhibitors. S glycoprotein is considered as the most important potential therapeutic target which is recognized by the cellular receptor and primed by the host cellular protease2. S1 and S2 are the two subunits of S protein; the S1 facilitates the entry of virus into the host cells by its binding to the host receptor through its receptor-binding domain (RBD), whereas S2 subunit facilitates the fusion of the viral and host membranes. The RBD in SARS-CoV-2 S protein was reported to be the gateway of infection that bound robustly to human angiotensin-converting enzyme 2 (ACE2) receptor3.
The SARS-CoV-2 genome-specific vaccines and their therapeutic antibodies are currently being tested. Alternatively, drug repurposing approaches of the existing therapeutic agents were carried out for clinical studies of COVID-19 therapeutics. Drugs such as remdesivir, favipiravir, hydroxychloroquine, ivermectin and lopinavir/ritonavir were repurposed for the treatment of COVID-19, based on their previous clinical history for potential therapeutics of other virus infections and pathologies4. These clinical therapies can be divided into two categories depending on their target: first, acting directly against SARS-CoV-2 either by inhibiting pivotal viral enzymes/proteins responsible for the replication of genomes or by preventing the entry of viruses into human cells; second, boosting the innate response by modulating the human immune system, this plays a key role against viruses, or by inhibiting the inflammatory processes that cause lung injury.
Based on the drug targets, clinical trials have been done on several classes of drugs including favipiravir and remdesivir (RNA polymerase inhibitors), lopinavir/ritonavir (protease inhibitors), chloroquine along with its hydroxyl derivative (aminoquinolines), xiyanping injection and corticosteroids (anti-inflammatory agents) and ACE2 inhibitors5. In addition to the clinical benefits of aminoquinolines, protease inhibitors and RNA polymerase inhibitors over COVID-19 therapeutics, there have been a few controversial findings reported from various research findings67.
Remdesivir was reported to inhibit SARS-CoV-2 which could be a prospective treatment of SARS-CoV-2, responsible for COVID-198. It was permitted to enter the clinical trials immediately under COVID-19 emergency conditions, on the basis of its safety and antiviral activities9. In an in vitro investigation, a hepatitis C virus protease inhibitor, namely simeprevir, was found to be a favourable repurposable drug for the treatment of COVID-19, which was also shown to synergize remdesivir in suppressing the replication of SARS-CoV-210.
Understanding the importance of RBD for COVID-19 therapeutics, limitations of vaccine development at a short interval, research controversies and prioritizing the urgent need of COVID-19 therapeutics, we performed an integrative computational approach to inhibit the RBD of S protein from SARS-CoV-2 using 56 drugs which were commercially available and used for therapeutics of various diseases. In this approach, an attempt was made to repurpose the drugs with potential antiviral activity against COVID-19 which could inhibit the RBD of S protein and ultimately prevent its entry into the human cells through ACE2 receptor.
Material & Methods
Sequence, structure and domain architecture analysis: The sequence, structure and functional information of RBD of S protein of SARS-CoV-2 were retrieved from National Center for Biotechnology Information (NCBI), GenBank with accession number MN908947. The spike (S) protein consisted of 1273 amino acids (aa), of which 229 were aa codes for RBD protein of SARS-CoV-2 that directly interacted with human ACE2. The RBD lies within the S1 region of a coronavirus S protein that triggers host-cell receptor binding activity11. The experimental structure of RBD with Protein Data Bank (PDB) ID: 6M0J was retrieved from Research Collaboratory for Structural Bioinformatics (RCSB), PDB with a resolution of 2.45 Å. The co-crystallized human ACE2 was removed using BIOVIA Discovery Studio 4.5 Visualizer (BIOVIA, San Diego, CA, USA).
Retrieval of drugs: The information of 56 commercially available drugs that are mostly prescribed for viral diseases, was retrieved from various sources6121314. The three-dimensional (3D) structure of all these 56 drugs was retrieved from the NCBI PubChem database in Structure Data Format, which was later converted to PDB format using online web server namely Simplified Molecular Input Line Entry System as per the requirement of AutoDock 4.2 docking tool (http://autodock.scripps.edu/).
Prediction of binding site: The key interacting residues were analyzed from the experimental structure of RBD-ACE2 complex (PDB ID: 6M0J) and available literature, which were identified as active site residues that take part in the binding site formation.
Molecular docking using AutoDock 4.2 tool: Molecular docking studies were carried out using experimental structure of RBD S protein against all the 56 drugs using AutoDock 4.2 tool. To allocate Kollman charges for the protein and Gasteiger partial charges for all the inhibitors, ADT v.1.5 was used. The grid with dimension space and parameters were as follows: x-centring: -37.872, y-centring: 28.878 and z-centring: 2.979, were chosen to allow full-extended conformation of the ligand. Based on the binding energy values, ligand efficiency, intermolecular hydrogen (H)-bonds and other hydrophobic and electrostatic interactions, the best resulting docked complexes were identified and processed for further computational analysis. The existence of intermolecular interactions between RBD–drug complexes was depicted through LigPlot+ (https://www.ebi.ac.uk/thornton-srv/software/LigPlus/).
Quantum chemical calculation using density functional theory (DFT): A density functional theory (DFT)-based study using the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy was carried out to investigate the reactivity and efficiency of potential drugs with antiviral activity against RBD by concerning the Becke, 3-parameter, LeeYang-Parr (B3LYP) correlation function of DFT15. The energy calculations were made using ORCA Program version 4.016. The electronic energy, frontier HOMOs, LUMOs, gap energy and dipole moment were calculated for the potential drugs. The DFT was calculated using the following equation:
E = minn {∫ Vnuclei (r̄)n (r̄)d3r̄ + F[n (r̄]}
Where, Vnuclei+n r }n ≡ trial density and F ≡ universal functional.
Molecular dynamics (MD) simulations: GROMOS96 43A1 force field in the GROMACS version 5.1.4 molecular dynamics (MD) simulation package was used to analyze the Apo (protein only) and Holo (protein-ligand) states to understand the dynamic behaviour, binding mode and specificity of RBD inhibitors. RBD-simeprevir complex, which represented the highest binding affinity from our docking analysis, was further processed for MD simulations to explore its inhibitor specificity, dynamic behaviour and mode of binding activity. The 'pdb2gmx' programme of GROMACS package was used for the generation of topology file. The steepest descent method with a 1000 kJ/mol tolerance was used for energy of minimization as required for releasing the conflicting interactions. In the primary phase, constant number of particles, volume and temperature (NVT) ensemble was carried for temperature equilibration by restraining the positions of backbone atoms for 1000 picoseconds (psec) followed by the secondary phase where pressure equilibration was used in constant number of particles, pressure and temperature (NPT) ensemble for 1000 psec. A run of 30 nanoseconds (nsec) MD time period was set for both Apo and Holo states using periodic boundary conditions with constant temperature. Visual MD (VMD 1.9.1) was used to analyze the resultant trajectories which are inbuilt in GROMACS. The root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), total energy and solvent accessible surface area (SASA) were analyzed using gmx_rmsd, gmx_rmsf, gmx_gyrate, gmx_tenergy and gmx_sasa. All 2D plots were graphed using GRaphing, Advanced, Computation and Exploration 5.1.23 version (https://www.its.hku.hk/services/research/hpc/software/grace) for data analysis of MD simulations.
Principal component analysis (PCA): To recognize the coordinated movements in Apo and Holo states of RBD, principal component analysis (PCA) was performed through essential dynamics (ED) process using gmx_covera and gmx_aneig tools as per the protocol within the software package of GROMACS. After diagonalizing and calculating the covariance matrix representing the molecules' concerted motion, a set of eigenvectors and eigenvalues were obtained.
Results
Annotation of binding site of RBD of spike protein: The literature survey and crystal structure analysis of RBD–ACE2 complex denoted the residues Lys417, Gly446, Tyr449, Tyr453, Leu455, Ile472, Phe486, Asn487, Tyr489, Gln493, Gly 496, Thr500, Asn501, Gly502 and Tyr505 of RBD, which interact with human ACE2 during cell entry, thus annotated as active site residues that take part in binding site formation17.
Molecular docking: The binding free energies of all the 56 (RDB-drug complexes) interactions are summarized in Table I. The docking results reflected different binding free energies for different drug-target complexes, ranging from −0.23 to −8.52 kcal/mol. RBD-chloroquine phosphate docking complex represented the least binding energy, whereas RBD-simeprevir complex represented the highest binding affinity among the 56 drugs under investigation. The drugs remdesivir and favipiravir represented the binding energy −4.68 and −4.32 kcal/mol which were lower than simeprevir. To identify the potential drugs with better antiviral activity for SARS-CoV-2 therapeutics, a cut-off binding energy with a range from −5.98 to −8.52 kcal/mol was considered for further computational analysis. The selected range provided eight drugs consisting of chalcone, grazoprevir, enzaplatovir, dolutegravir, daclatasvir, tideglusib, presatovir and simeprevir. The drugs bortezomib, entecavir, ribavirin and trifluridine were considered due to the highest number of conventional H-bond interactions with RBD. Remdesivir was also considered under priority research area for COVID-19 therapeutics. Similarly, the drug favipiravir was considered for further analysis based on its applications for COVID-19 therapeutics.
| PubChem Compound ID | Drug | Binding energy (kcal/mol) | Ligand efficiency | Inhibition constant (μm) | Number of H-bonds | H-bond forming residues | Average distance of H-bonds (Å) |
|---|---|---|---|---|---|---|---|
| 24873435 | Simeprevir | −8.52 | −0.16 | 564.68 | 4 | GLN493, SER494 | 2.810 |
| 58029842 | Presatovir | −6.92 | −0.19 | 8.42 | 4 | TYR449, GLN498, GLY496 | 2.727 |
| 11313622 | Tideglusib | −6.81 | −0.28 | 10.27 | 1 | ARG403 | 3.088 |
| 58406357 | Enzaplatovir | −6.81 | −0.24 | 10.21 | 1 | GLY496 | 3.074 |
| 44603531 | Grazoprevir | −6.46 | −0.12 | 18.41 | 1 | GLY496 | 2.782 |
| 25154714 | Daclatasvir | −6.38 | −0.12 | 20.98 | 3 | GLN493, SER494, GLY502 | 2.827 |
| 54726191 | Dolutegravir | −6.36 | −0.21 | 21.8 | 5 | TYR449, GLN493, SER494 | 2.862 |
| 637760 | Chalcone | −5.98 | −0.37 | 41.28 | 1 | GLY496 | 2.822 |
| 5284373 | Cyclosporine A | −5.93 | −0.07 | 44.68 | 4 | TYR449, TYR453, GLY496 | 2.619 |
| 72193873 | Interferon-alpha | −5.89 | −0.29 | 48.37 | 1 | TYR453 | 1.887 |
| 5281040 | Montelukast | −5.87 | −0.14 | 49.62 | 1 | GLU406 | 1.843 |
| 135398508 | Entecavir | −5.81 | −0.29 | 55.18 | 8 | ASN487, GLU484, GLY485, CYS488, PHE490, GLN493 | 2.179 |
| 5277135 | Elvitegravir | −5.71 | −0.18 | 65.57 | 2 | ARG403, GLU406 | 2.463 |
| 54671008 | Raltegravir | −5.62 | −0.18 | 76.12 | 2 | GLY502, TYR453 | 2.701 |
| 193962 | Etravirine | −5.6 | −0.2 | 78.6 | 1 | TYR449 | 3.343 |
| 441243 | Saquinavir | −5.46 | −0.11 | 99.15 | 2 | THR500 | 1.943 |
| 64139 | Efavirenz | −5.45 | −0.26 | 101.27 | 3 | TYR449, GLN498 | 2.692 |
| 3194 | Ebselen | −5.45 | −0.34 | 101.16 | 1 | ARG403 | 2.714 |
| 11285588 | Danoprevir | −5.37 | −0.11 | 116.51 | 4 | GLN498, THR500, ASN501 | 2.688 |
| 5316606 | Deoxyrhapontin | −5.26 | −0.18 | 138.29 | 5 | TYR449, GLN498, TYR449, GLY496, TYR505 | 2.350 |
| 4124851 | Tdzd-8 | −5.2 | −0.35 | 154.31 | 1 | ARG403 | 3.214 |
| 5362440 | Indinavir | −5.19 | −0.12 | 158.04 | 2 | SR494, TYR505 | 2.549 |
| 64143 | Nelfinavir | −5.14 | −0.13 | 170.4 | 4 | GLN493, TYR553, GLN493, SER494 | 2.511 |
| 2577 | Carmofur | −5.12 | −0.28 | 178.07 | 5 | GLN493, GLY496, GLN498 | 2.736 |
| 441300 | Abacavir | −5.11 | −0.24 | 180.12 | 3 | YTR453, GLN493, GLU406 | 2.093 |
| 387447 | Bortezomib | −5.09 | −0.18 | 186.84 | 6 | GLY496, GLN498, ASN501, THR500 | 2.209 |
| 479503 | Shikonin | −4.96 | −0.24 | 230.54 | 2 | SER494, GLN498 | 2.102 |
| 54682461 | Tipranavir | −4.95 | −0.12 | 234.88 | 3 | GLN493, SER494 | 2.626 |
| 24798764 | Lomibuvir | −4.86 | −0.16 | 273.36 | 1 | ARG403 | 3.395 |
| 10324367 | Boceprevir | −4.85 | −0.13 | 277.08 | 5 | ARG403, ASN501, TYR505 | 2.607 |
| 135398513 | Acyclovir | −4.82 | −0.3 | 290.89 | 5 | GLU484, TYR489, PHE490, GLN493 | 2.181 |
| 471161 | Maribavir | −4.74 | −0.2 | 333.77 | 5 | TYR449, GLY496, TYR553, GLN493 | 2.632 |
| 121304016 | Remdesivir (Investigational drug: drug already in use) | −4.68 | −0.11 | 371.48 | 4 | TYR449, GLN493, SER494 | 2.907 |
| 37542 | Ribavirin | −4.6 | −0.27 | 425.74 | 8 | ARG403, TYR449, GLY496, GLN498, TYR453 | 2.699 |
| 2719 | Chloroquine (Investigational drug: drug already in use) | −4.6 | −0.21 | 427.1 | 1 | ASN501 | 2.916 |
| 213039 | Darunavir | −4.42 | −0.12 | 572.51 | 4 | GLN498, THR500, ASN501 | 2.364 |
| 131411 | Arbidol (Investigational drug: drug already in use) | −4.33 | −0.15 | 668.95 | 1 | TYR453 | 3.535 |
| 492405 | Favipiravir (Investigational drug: drug already in use) | −4.32 | −0.39 | 678.1 | 4 | ARG403, GLY496, TYR453 | 2.659 |
| 5475158 | Cinanserin | −4.28 | −0.18 | 725.12 | 2 | GLN498, ASN501 | 2.067 |
| 11556711 | Carfilzomib | −4.21 | −0.08 | 819.77 | 2 | TYR453 | 2.465 |
| 219104 | Px-12 | −4.05 | −0.37 | 1.07 | 1 | SER494 | 1.920 |
| 60825 | Lamivudine | −3.92 | −0.26 | 1.34 | 2 | GLY496, GLU406 | 2.613 |
| 5281718 | Polydatin | −3.9 | −0.14 | 1.39 | 4 | ASN501, TYR449, GLY496, TYR505 | 2.243 |
| 135398748 | Penciclovir | −3.7 | −0.21 | 1.94 | 4 | SER494, GLN493, TYR453 | 2.138 |
| 92727 | Lopinavir (Investigational drug: drug already in use) | −3.49 | −0.08 | 2.76 | 2 | GLY496, TYR505 | 2.400 |
| 3652 | Hydroxy chloroquine (Investigational drug: drug already in use) | −3.42 | −0.15 | 3.1 | 3 | ARG403, TYR453 | 2.797 |
| 148192 | Atazanavir | −3.42 | −0.07 | 3.14 | 4 | TYR453, GLN493, | 2.390 |
| 16076883 | Asunaprevir | −3.4 | −0.07 | 3.2 | 3 | ARG403, GLN493, ASN501 | 3.075 |
| 3117 | Disulfiram | −3.29 | −0.21 | 3.87 | 1 | ASN501 | 2.884 |
| 6256 | Trifluridine | −2.99 | −0.15 | 6.46 | 6 | TYR449, GLY496, GLN498 | 2.817 |
| 135398740 | Ganciclovir | −2.97 | −0.17 | 6.71 | 5 | GLY496, GLN498, TYR505 | 2.177 |
| 447043 | Azithromycin | −2.57 | −0.05 | 13.06 | 1 | GLN493 | 2.807 |
| 3010818 | Telaprevir | −2.42 | −0.05 | 16.9 | 3 | GLY496, ASN501, GLN493 | 2.662 |
| 392622 | Ritonavir (Investigational drug: drug already in use) | −1.75 | −0.04 | 52.57 | 2 | TYR453, GLN493 | 3.181 |
| 131536 | Fosamprenavir | −1.4 | −0.04 | 94.32 | 2 | GLN498, ASN501 | 2.057 |
| 64927 | Chloroquine phosphate | −0.23 | −0.05 | 675.48 | 5 | LYS458, GLU471 | 2.180 |
Hydrogen bond analysis and inter-atomic distance calculation: The study showed that all docked complexes exhibited variable numbers of intermolecular H-bonding patterns. The docking analysis depicted eight H-bonds (average of ~2.179 and ~2.699 Å) in RBD complex with entecavir and ribavirin, which were the highest number among all the complexes (Fig. 1A and B). Remdesivir and favipiravir represented four H-bonds (average of ~2.907 and ~2.659 Å) (Fig. 1C and D). H-bond network of RBD-simeprevir complex resulted in four number of H-bonds in spite of highest binding affinity among all drugs. The H-bond network was between Lys417, Gln493 and Ser494 of RBD and simeprevir with an average distance of ~2.810 Å (Fig. 2A). The residues Tyr453, Leu455 and Lys417 were involved in hydrophobic interactions. The post-MD docking studies of RBD-simeprevir complex represented a binding affinity of −8.74 kcal/mol. The H-bond network was between Gly496 and Ser494 of RBD and simeprevir with an average distance of ~3.12 Å.
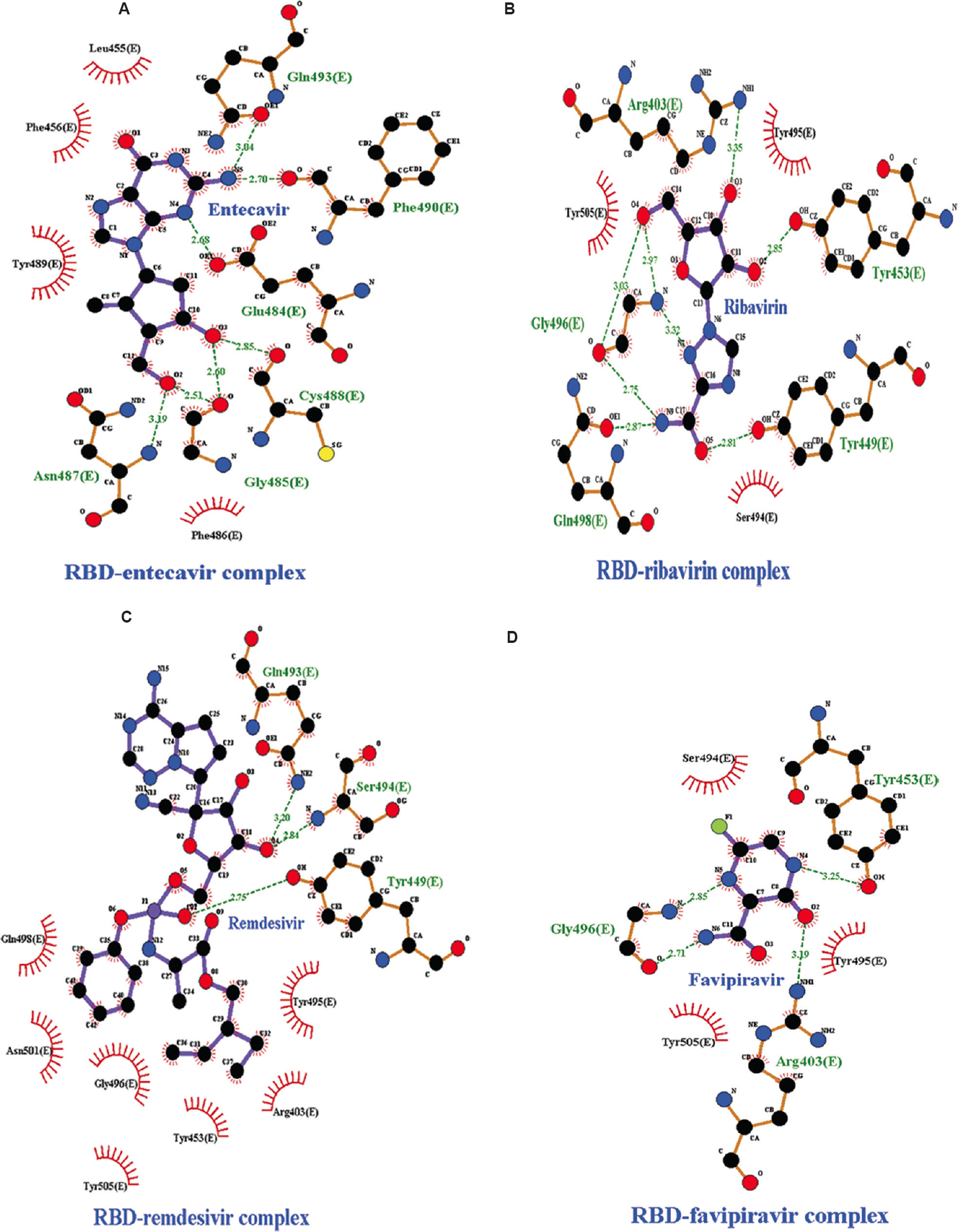
- Intermolecular hydrogen bonding, electrostatic and hydrophobic interactions formed between (A) RBD-entecavir complex, (B) RBD-ribavirin complex, (C) RBD-remdesivir complex, (D) RBD-favipiravir complex. The images are drawn by LigPlot+ tool. RBD, receptor-binding domain.
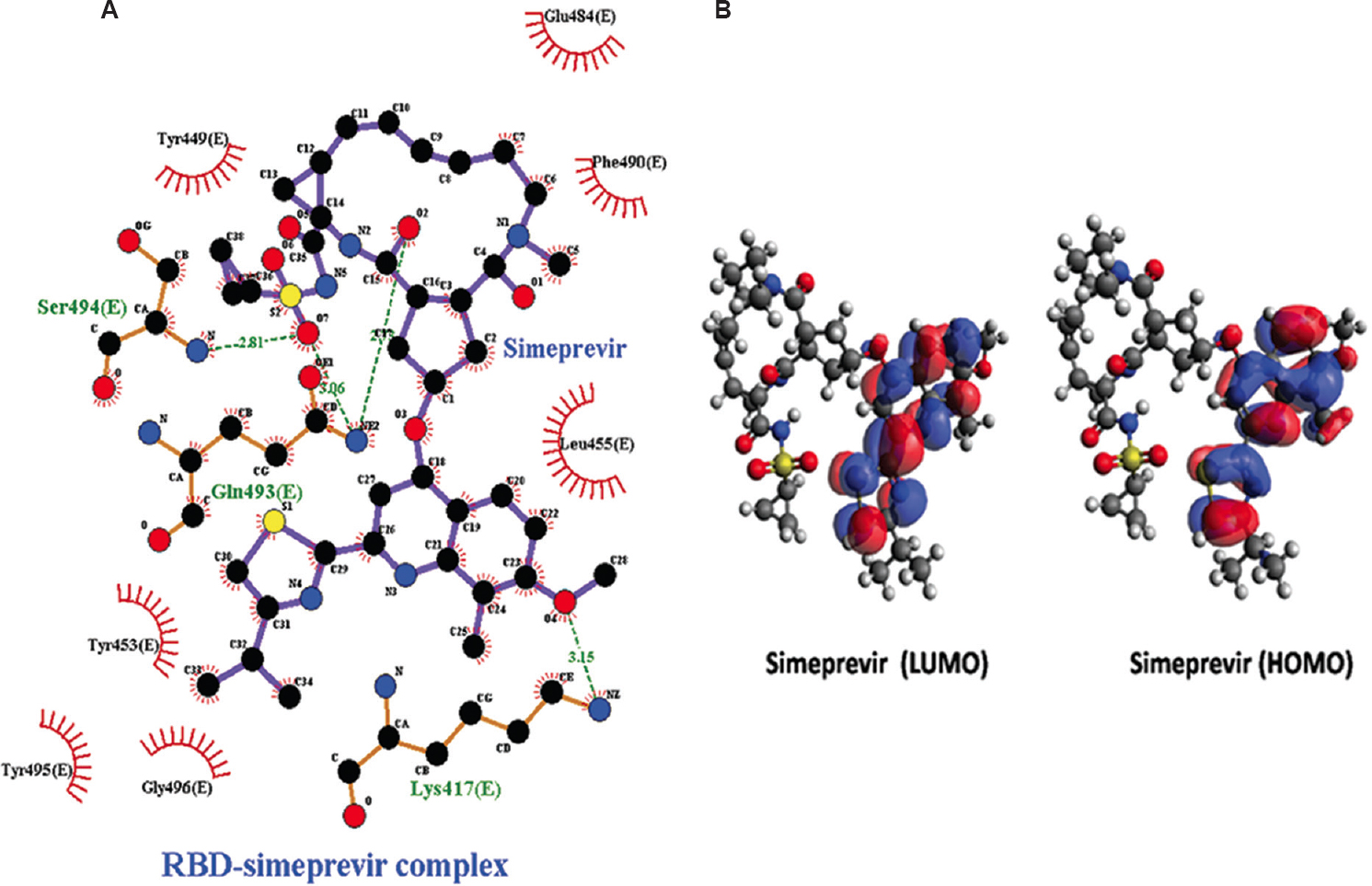
- (A) Intermolecular hydrogen bonding, electrostatic and hydrophobic contacts formed between RBD-simeprevir complex drawn by and LigPlot+ tool. (B) LUMO and HOMO plots of simeprevir which exhibited higher reactivity against RBD. The positive electron density is indicated by red colour while blue colour indicates negative electron density. HOMO, highest occupied molecular orbital; LUMO, lowest unoccupied molecular orbital.
Quantum chemical calculation: Owing to the importance of quantum computation, quantum chemistry was employed to study the frontier molecular descriptors such as HOMO and LUMO, gap energy and dipole moment for all the 14 shortlisted drugs that were predicted with better potential activity against RBD of S protein in SARS-CoV-2 (Table II). The effective reactivity for all the 14 drugs, which showed band energy gap (ΔE), i.e. the difference between ELUMO and EHOMO, ranged from 9.254 to 13.126 kcal/mol. Simeprevir displayed greatest reactivity against RBD among all the screened drugs based on its lowest band energy gap, which was calculated to be 9.254 kcal/mol (Fig. 2B). Taking together the results of molecular docking and DFT analysis, the drug simeprevir was further postulated for MD simulation along with RBD.
| PubChem Compound ID | Drug name | Electronic energy (eV) | ELUMO (kcal/mol) | EHOMO (kcal/mol) | GAPEnergy (ΔE) (kcal/mol) | Dipole Moment (Debye) |
|---|---|---|---|---|---|---|
| 37542 | Ribavirin | −59709.776 | 2.749 | −10.377 | 13.126 | 6.74289 |
| 6256 | Trifluridine | −78658.337 | 1.971 | −10.384 | 12.355 | 7.65268 |
| 135398508 | Entecavir | −69756.220 | 3.179 | −8.282 | 11.461 | 11.04225 |
| 58406357 | Enzaplatovir | −106784.442 | 2.502 | −8.712 | 11.214 | 1.32310 |
| 121304016 | Remdesivir (investigational drug: drug already in use) | −211558.873 | 2.972 | −8.100 | 11.072 | 11.35974 |
| 492405 | Favipiravir (investigational drug: drug already in use) | −32705.803 | 1.346 | −9.385 | 10.731 | 5.84279 |
| 54726191 | Dolutegravir | −119111.197 | 1.640 | −8.971 | 10.611 | 7.00435 |
| 387447 | Bortezomib | −103013.589 | 1.610 | −8.525 | 10.135 | 9.27791 |
| 637760 | Chalcone | −42552.894 | 1.362 | −8.672 | 10.034 | 3.48279 |
| 44603531 | Grazoprevir | −300982.758 | 1.466 | −8.414 | 9.88 | 6.97561 |
| 25154714 | Daclatasvir | −256640.071 | 1.678 | −8.186 | 9.864 | 7.18234 |
| 11313622 | Tideglusib | −94283.082 | 1.596 | −8.090 | 9.686 | 2.53781 |
| 58029842 | Presatovir | −184960.093 | 2.174 | −7.114 | 9.288 | 4.29299 |
| 24873435 | Simeprevir | −297899.418 | 1.436 | −7.818 | 9.254 | 5.44166 |
Trajectory analysis of MD simulations: The MD simulation of the Apo and Holo (RBD-simeprevir complex) states of RBD was carried out to evaluate the dynamics and stability of RBD protein, RMSD, Cα-RMSF, Rg, total energy and SASA from the trajectories resulted from MD simulations using GROMACS tools. The RMSD depiction for backbone residues was developed and plotted against a time scale of 30 nsec to access the dynamic stability of RBD. The backbone RMSD (Holo) was observed with a stable deviation after 20 nsec of simulation (Fig. 3A) when compared to its Apo state. The Apo state depicted a significant deviation from 0 to 30 nsec (0.1-0.25 nm) compared to the Holo state, with a stable RMSD with a value ranging from ~0.23 to ~0.22 nm from 25 to 30 nsec. This means that the drug simeprevir can help stabilize the protein by changing the conformation. The result of RMSD was further validated through fluctuation of residues using RMSF. The mobility of different residues was observed in RBD through RMSF plots (Fig. 3B). In Apo state, it was observed that the amino acid residues between 370-379 and 430-435 exhibited greater deviations in their Cα atoms in comparison to other regions. Around 10 residues (475-485) displayed greater deviations in Holo state of RBD as compared to its Apo state. This plot signifies that binding of simeprevir decreases the mobility of residues in Holo state than in Apo state. Rg was calculated to analyze the overall compactness for both the states. Gyration radius versus time graphs were plotted to check the compactness (Fig. 3C). The Apo state's Rg ranged from ~1.84 to ~1.85 nm, whereas the Holo Rg ranged from ~1.82 to ~1.79 nm, which represented higher Rg value in Apo state than in Holo state. The energy plot depicted the decreased mobility of residues in Holo state compared to Apo state, which was confirmed form RMSF plot (Fig. 3D). The hydrophobic interactions mediate the exposure of amino acids to certain solvent. The frequency of these interactions with the solvent and the core protein residues is directly proportional to the exposed surface area. The sketch of SASA (Fig. 3E) showed a reduction in the accessible solvent surface in the Holo state of RBD relative to its Apo state. SASA's findings showed the alteration of hydrophilic and hydrophobic interaction areas resulted by the binding of simeprevir to RBD, which could potentially prevent the host-viral interactions and ultimately making binding surface unavailable for the virus with human counterparts. Throughout the simulation time, the SASA graphs of the Holo state represented SASA with ~91 to ~110 nm2, which was lower than the Apo state with a value of ~115 to ~ 120 nm2 (Fig. 3E).
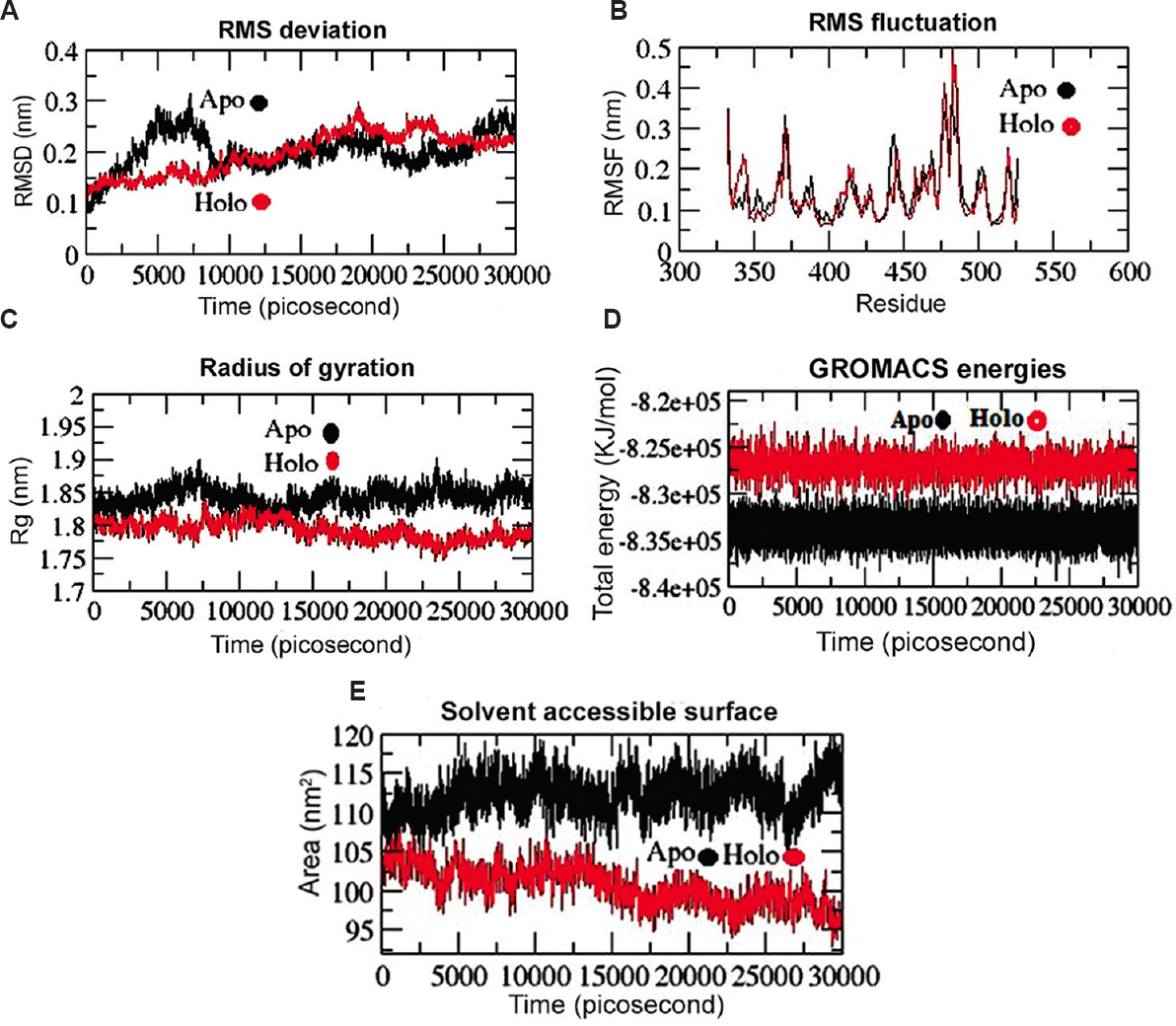
- Conformational stability of RBD (Apo and Holo states) from SARS-CoV-2 spike protein throughout 30 nanoseconds (nsec) time period of MD simulations. (A) Backbone-RMSD of RBD. (B) Cα-RMSF profile of RBD. (C) Radius of gyration (Rg) profile of RBD. (D) Total energy of RBD and RBD-simeprevir complex (Apo and Holo state) during 30 nsec MD simulations. (E) Solvent accessible surface (SASA) analysis of RBD-simeprevir complex during 30 nsec MD simulations. The Apo and Holo are displayed by black and red lines, respectively. RMSD, root mean square deviation; RMSF, root mean square fluctuation; MD, molecular dynamics.
H-bond analysis: The intermolecular hydrogen bonds of the RBD-simeprevir complex were tracked using the gmx_hbond tool of GROMACS (Fig. 4A). The simulation of Holo state represented an inconsistent number of intermolecular hydrogen bonds throughout the simulation time period. It represented four H-bonds (with an average atomic distance of ~1.67 nm). The number of H-bonds was directly proportional to the stability of the drug-target complex over the entire simulation time period. During simulations, a few crucial H-bonds such as Gln493 and Lys417 were broken, but later novel H-bonds, van der Waals and hydrophobic contacts were compensated (Fig. 4B). In spite of certain novel interactions with residues Leu452, Phe 456, Tyr489, Leu492 and Tyr505, it did not compensate with residue Ser494 (H-bond). This reflects its potentiality against the targeted protein as Ser494 which is one of the crucial residues in boosting the ACE2 binding18.

- (A) Deviation of H-bonds contributed in interaction during 30 nsec simulation in RBD-simeprevir complex. (B) Post-MD simulations intermolecular hydrogen bonding, electrostatic and hydrophobic contacts formed between RBD-simeprevir complex drawn by Diglot+ tool. (C) The cloud represents the projection of trajectories eigenvectors (EV1 and EV2) (Black: Apo; Red: Holo). (D) Projection of the motion of the Apo and Holo states of RBD in phase space along the first two principal eigenvectors (EV1 and EV2).
Principal component analysis (PCA): The movement of the RBD Apo and Holo states in phase space was captured by the trajectory projections from PC1 and PC2 (Fig. 4C), which were well aligned with RMSF (Fig. 3B). The trace values shown for Apo and Holo were 13.65 and 12.31 nm2 for RBD. The lower Holo state trace values confirmed the overall decrease in RBD flexibility relative to its Apo state. The motion direction was shown by the vectorial representation of the solitary components. The majority of internal movements are shown by the initial vectors, while EV1 and EV2 represent a large number of overall movements. Following the plotting of eigenvalues against eigenvectors, steep curves of eigenvalues (Fig. 4D) were obtained.
Discussion
Attempts for the development of vaccines and direct-acting potential antiviral drugs are being carried out for effective treatment of COVID-19 therapeutics. Existing reports from various research findings have suggested that drugs such as chloroquine, hydroxychloroquine, arbidol, remdesivir, favipiravir, azithromycin and nelfinavir have shown efficacy and safety for COVID-19 treatment121314. Pharmaceutical and medicinal experts, however, raised the queries on their efficacy because both these drugs were originally designed to treat other diseases. This depicted their implementation through drug-repurposing. Considering the pros and cons of the COVID-19 investigational drugs, the current investigation was based on in silico screening of 56 commercially available drugs against RBD, through a computational approach to find out potential candidates for COVID-19 therapy. This investigation revealed chloroquine phosphate and simeprevir to have the least and highest antiviral activity based on their binding energy. RBD-simeprevir complex, which was approached for MD simulation predicted higher fluctuation pattern in a few residues of RBD, which might be due to their direct interaction with human ACE2 as reported by Wu et al1. The integrative computational approach of docking, quantum chemical calculation and MDS was used, which was able to detect the linkage between residual movements in RBD and its way of interactions with these drugs. The capability of simeprevir for COVID-19 therapeutics was eventually hypothesized by the Apo and Holo state trajectories. The overall findings obtained from various computational tools affirmed the pivotal role of the 14 drugs. This investigation was an in silico approach based on the information of drugs and experimentally derived crystal structure of RBD. The findings of this in silico investigation could be a supporting evidence for in vivo and in vitro studies needed to be carried out to confirm the efficacy and antiviral drug potency of simeprevir against RBD SARS-CoV-2 S protein.
Acknowledgment:
Authors thank Prof. (Dr) Balram Bhargava, Secretary, Department of Health Research, Ministry of Health and Family Welfare Government of India, and Director-General, Indian Council of Medical Research, New Delhi, for his support.
Financial support & sponsorship: None.
Conflicts of Interest: None.
References
- A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265-9.
- [Google Scholar]
- SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271-80.
- [Google Scholar]
- Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol. 2020;17:613-20.
- [Google Scholar]
- A review of SARS-CoV-2 and the ongoing clinical trials. Int J Mol Sci. 2020;21:2657.
- [Google Scholar]
- Available evidence and ongoing clinical trials of remdesivir: Could it be a promising therapeutic option for COVID-19? Front Pharmacol. 2020;11:791.
- [Google Scholar]
- Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int J Antimicrob Agents. 2020;56:105949.
- [Google Scholar]
- Efficacy of hydroxychloroquine in patients with COVID-19: Results of a randomized clinical trial. medRxiv 2020 doi: 10.1101/2020.03.22.20040758
- [Google Scholar]
- Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269-71.
- [Google Scholar]
- Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. 2018;9:e00221-18.
- [Google Scholar]
- Simeprevir suppresses SARS-CoV-2 replication and synergizes with remdesivir. bioRxiv 2020 doi: 10.1101/2020.05.26.116020
- [Google Scholar]
- Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260-3.
- [Google Scholar]
- Discovering drugs to treat coronavirus disease 2019 (COVID-19) Drug Discov Ther. 2020;14:58-60.
- [Google Scholar]
- Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput Struct Biotechnol J. 2020;18:784-90.
- [Google Scholar]
- Nelfinavir was predicted to be a potential inhibitor of 2019-nCoV main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. bioRxiv 2020 doi: 10.1101/2020.01.27.921627
- [Google Scholar]
- The performance of the Becke-Lee-Yang-Parr (B-LYP) density functional theory with various basis sets. Chem Phys Lett. 1992;197:499-505.
- [Google Scholar]
- Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215-20.
- [Google Scholar]
- Structural proteins in severe acute respiratory syndrome coronavirus-2. Arch Med Res. 2020;51:482-91.
- [Google Scholar]




