Translate this page into:
Spinocerebellar ataxia 7 (SCA7) in Indian population: predilection of ATXN7-CAG expansion mutation in an ethnic population
Reprint requests: Dr Achal Kumar Srivastava, Department of Neurology, Neurosciences Centre, All India Institute of Medical Sciences, Ansari Nagar, New Delhi 110 029, India e-mail: achalsrivastava@hotmail.com
-
Received: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
Spinocerebellar ataxia 7 (SCA7) is a rare form of neurodegenerative disorder with the clinical manifestation of cerebellar ataxia and retinal degeneration. In this study we describe the clinico-genetic characteristics of nine SCA7 families of Indian origin and cross compare these with other available worldwide studies.
Methods:
Thirty five individuals from nine SCA7 families were clinico-genetically characterized and CAG repeat distribution analysis was carried out in 382 control DNA samples from healthy controls (derived from 21 diverse Indian populations based on ethnic and linguistic and geographical location).
Results:
Of the nine families studied, 22 affected individuals and one asymptomatic carrier were identified. The average age at disease onset was 23.4±12.6 yr. The length of expanded CAG ranged from 40-94 with mean value of 53.2±13.9. The main clinical findings in affecteds individuals included cerebellar ataxia, and retinal degeneration along with hyper-reflexia (95%), slow saccades (85%) and spasticity (45%). Analysis of the association of number of CAG repeats with disease onset revealed that<49 repeats were associated with earlier age at onset in South East Asians compared to European populations. Further analysis of CAG repeats from 21 diverse Indian populations showed pre-mutable repeats (28-34) alleles in the IE-N-LP2 population. Six of the nine families identified in this study belonged to the same ethnic population.
Interpretations & conclusion:
Our results show that presenece of SCA7 is relatively rare and confined to one ethnic group from Haryana region of India. We observed a homogeneous phenotypic expression of SCA7 mutation as described earlier and an earlier age of onset in our patients with CAG <49. The identification of pre-mutable allele in IE-N-LP2 suggests this population to be at the risk of SCA7.
Keywords
CAG repeats
genotype-phenotype correlation
Indian population
SCA7
spinocerebellar ataxia

The autosomal dominant cerebellar ataxia-II (ADCA-II) presents cerebellar ataxia and retinal degeneration as the clinical symptoms1. Spinocerebellar ataxia (SCA7) is the only disorder recognized from this group till date and the CAG repeat expansion in exon 3 of ATXN7 (in the region of chromosome 3p12-p21.1) has been reported to be the causal mutation2. CAG repeats range bewteen 4-18 in healthy individuals, although repeat length of 28-33 is also regarded as mutable normal alleles. Repeats of 34 and 35 are alleles with reduced penetrance and are of questionable significance. Repeats in 37-460 range have been described in affected individuals and are the pathogenic full-penetrance alleles345. The SCA7 mutation has been identified in various ethnic groups and geographical regions around the world but the prevalence of SCA7 has been shown to be the highest among cases of familial ADCAs in South Africa (22%)6 and 55 per cent among 27 genetically confirmed SCAs in the Scandinavian region7. The SCA7 phenotypes results from neuro-ophthalmic degeneration. Apart from cerebellar ataxia and pigmentary retinal degeneration, slowing of saccades and hyperreflexia have been found to be associated in the majority of the cases1389. The onset of cerebellar ataxia and retinal degeneration varies with the number of CAG repeats; higher repeats are associated with increasing severity and early onset. The length of the expanded repeats also affects the symptom presented at the onset of the disease and during its progression. There is a correlation between the increase in CAG repeat length and the chronological deterioration of nervous system. Diminution of the vision precedes cerebellar ataxia with higher repeat length and vice versa with lower repeat sizes310. The prevalence of SCA7 is largely determined by local founders, although the haplotype is not shared among the identified SCA7 families studied in the population which is indicative of multiple and recurrent origin of SCA7 in various populations11. The prevalence of SCA7 in Indian population is very low1213. In an earlier study, we have reported two SCA7 families identified out of 235 clinically diagnosed families of SCAs in our cohort, where we had shown the different origin based on haplotype analysis using microsatellite markers14. Subsequently in the last seven years, in our Ataxia clinic at All India Institute of Medical Sciences, New Delhi, we have been successful in identifying seven more families of SCA7. The aim of the current study is to entail the clinico-genetic characterization of nine SCA7 kindred and their comparison to other reported SCA7 phenotypes. We also have carried out CAG repeat distribution analysis in 20 populations of diverse ethnicities of Indian origin to estimate the frequency of predisposed chromosomes for SCA7 mutation.
Material & Methods
This clinico-genetic study was conducted at the All India Institute of Medical Sciences (AIIMS), New Delhi and CSIR-Institute of Genomics and Integrative Biology (IGIB), Delhi, India. In this prospective observational study, a total of nine families with clinical and genetic diagnosis of SCA7 were identified out of 949 SCA families (both familial and non-familial history of disease; 402 families displayed autosomal dominant mode of disease inheritance) seen in the Ataxia clinic from 1997 to 2011, at the department of Neurology, AIIMS, New Delhi. All cases had undergone SCA repeat expansion mutation screening for SCA1-3, SCA6, SCA7, SCA8, SCA12 and DRPLA (dentatorubral-pallidoluysian atrophy) at the CSIR-IGIB, Delhi. The study protocol was approved by the institutional ethics committee of the participating institutes. Written informed consent was obtained from each participant.
Sample size and sample selection:
(i) SCA7 families - The relatives of all the index cases were called to participate in this study for performing the clinico-genetic evaluation of SCA7. A total of 35 members from nine families were included (comprising 22 affected, 13 unaffected individuals). The term affected refers to clinically symptomatic and mutation positive individuals; unaffected are neurologically normal individuals of the patient's family; and asymptomatic refers to clinically normal individuals who carry the mutation at the genetic level without any phenotypic manifestations. Two of these nine SCA7 families have already been described earlier14, but in this study, two more relatives in one family comprising four affected and seven unaffected individuals, were included. Seven new families (comprising 18 affected, five unaffected) were also included. One unaffected individual later refused to participate in this study. Six of the nine identified families shared a common ethnic background (IE-N-LP2) and hailed from Haryana, a State situated in the northern part of India; the remaining families also had north-Indian origin and were from large population groups (Indian Genome Variation consortium)15.
(ii) Healthy population controls - The CAG repeat length distribution analysis was carried out in DNA samples of 382 healthy individuals from 21 different ethnic Indian populations (repository of DNA samples from Indian Genome Consortium study15 at CSIR-IGIB), consisting of nine Indo-European (North India), ten Dravidian (South India) and one Tibeto-Burman origin. Additionally, we took 54 unrelated control DNA samples from IE-N-LP2 population from the repository of IGV consortium study (IE-N-LP2 population ethnically matched to most of our SCA7 families in the present study). The convention for population codes as described in the original study of IGV consortium15 was adhered to the codes are described as L(linguistic affiliation), G(geographical location) followed by E(ethnic category). Linguistic categories described are Indo-European (IE), Dravidian (DR), Austro-Asiatic (AA) and Tibeto-Burman (TB). Geographically, the population samples belong to Central (C), North (N), East (E), West (W) and North-East (NE) regions of India. The ethnic categories are Large caste Population (LP), Isolated tribal Population (IP) and Small religious Population (SP).
Neurological evaluation: All individuals were subjected to neurological examination either in the Ataxia clinic or at their homes. The information about the age at onset of various symptoms was obtained by direct interview with the patient and their family informants. The affected individuals were assessed for ataxia severity using Scale for the Assessment and Rating of Ataxia (SARA)16. The ophthalmological evaluation was carried out with visual acuity tests, field of vision, fundoscopic examination and electroretinography wherever possible. For 11 patients CT-head/MRI-brain images were obtained for radiological assessment of brain. All families except one were periodically assessed in follow up for symptom evolution in the clinic.
CAG repeat analysis of ATXN7 gene: Peripheral venous blood (8 ml) was collected from each participant in the Ataxia clinic. DNA was isolated by salting out method. The repeat length estimation was carried out initially by PCR amplification using primer 6 FAM- (5’ AAGGAGCGGAAAGAATGTCG 3’) and 4U716 (5’ CACGACTGTCCCAGCATCACTT 3’) following standard procedure2. The labelled PCR products were analyzed by fragment analysis on ABI 3130xl automated sequencer (ABI Foster City, USA). The confirmation of exact repeat size was done by PCR amplification of repeat region using primers 4U1024 and 4U716 followed by direct sequencing with ABI sequencing kit and then analyzed on ABI 3130xl automated sequencer (ABI Foster City, USA).
Statistical analysis: The association between CAG repeat length and age at onset was performed by Pearson's correlation. The difference of means between two groups was computed by Student's t test.
Results
General demographic characteristics of SCA7 families: The expansion of CAG repeats in ATXN7 beyond the pathological threshold was identified in 22 patients from nine different families. The frequency of SCA7 among the families with autosomal dominant inheritance of cerebellar ataxias was 2.2 per cent (9/402 families). The pathogenic repeat expansion at other known SCA loci (SCA1, SCA2, SCA3, SCA6, SCA8, SCA12 and DRPLA) was excluded in SCA7 affected individuals. The average age at examination of the 22 affected individuals was 30.6±15.4 (range, 5-58) years, average age at disease onset was 23.4±12.6 (range, 4-49) yr with male:female ratio of 14:8. The length of expanded CAG alleles ranged from 40-94 with mean±SD 53.2±13.9 (Table I). The detailed clinical observations of 22 mutation carriers are described in Table II. One 12 year old female child (AT2126) was an asymptomatic carrier of the SCA7 mutation (CAG51) and was not included for clinical data analysis. Except for the four affected, all other patients had cerebellar ataxia and vision impairment together (Table II). Two individuals were not available for clinical evaluation and their disease history was obtained telephonically and by direct conversation with family informants. In the remaining 20 patients, the frequency distribution of a variety of clinical features showed prominent features like gait ataxia and visual impairment (95.45 and 86.4%, respectively), however, hand incoordination was observed in all affected individuals. Among other related neurological associations, brisk reflexes, slow saccades and extensor plantar responses were present in majority of the patients. Spasticity and ophthalmoplegia were observed in moderate frequency among patients (Table III).
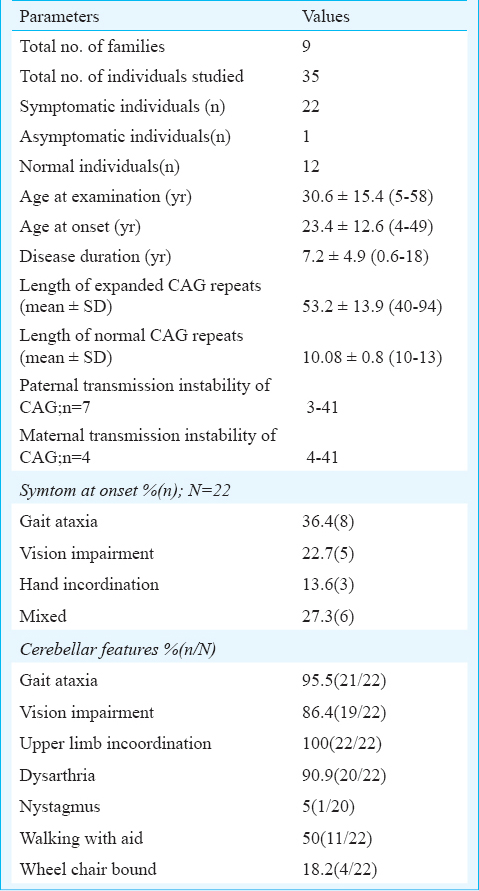
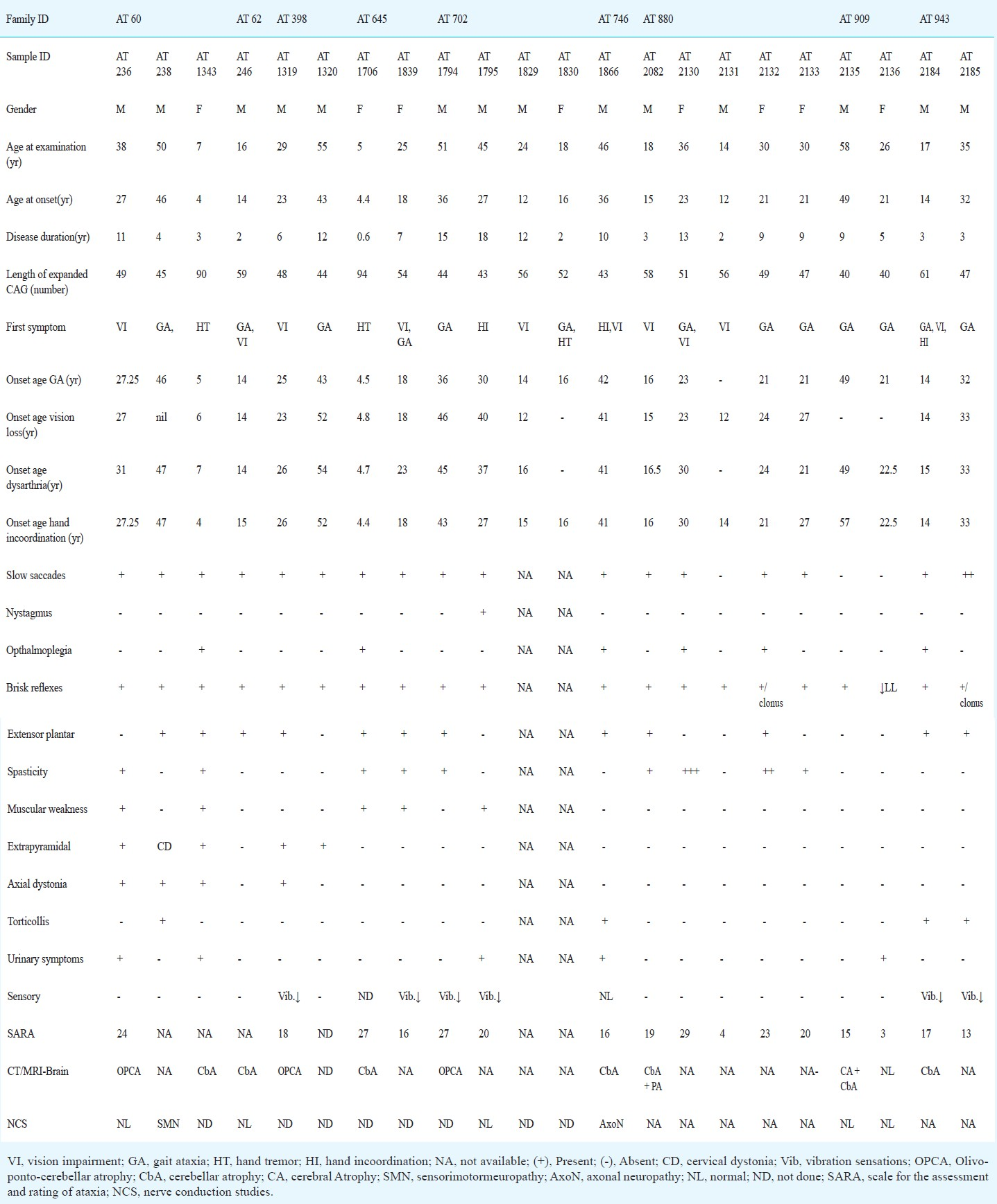
Paraclinical abnormalities in SCA7 patients: Among the paraclinical investigations performed (Table II), CT head/MRI-brain could be obtained for 11 patients and for 10 patients findings were consistent with olivopontocerebellar atrophy (OPCA) or cerebellar atrophy (Fig. 1). Electrophysiological studies conducted in seven patients showed no abnormality except for two patients where each showed a pattern suggestive of sensory-motor neuropathy and axonal neuropathy, respectively. Visual evoked potential (VEP) and brain stem auditory evoked response (BAER) studies conducted in four patients showed abnormality in two patients (CAG-49 and 43) and two had no impairment (CAG-40 each). Detailed fundus examination was carried out for seven affected individuals. One patient was (AT1839) found to have no gross changes in bilateral eyes despite subjective complaint of diminution of vision, the remaining patients showed degenerated retinal pigmentary epithelium (RPE) at the macular region (Fig. 2A–C.). Electroretinography recording was done for three patients including AT1839 and all showed abnormalities like extinguished response (AT236), reduced B-wave amplitude in Rt/Lt eyes 38.06/38.09 μV (AT1794) and mildly reduced B-wave amplitude in right eye-57.54/Lt eye-60.11 μV (AT1839) (Fig. 2D–F.).
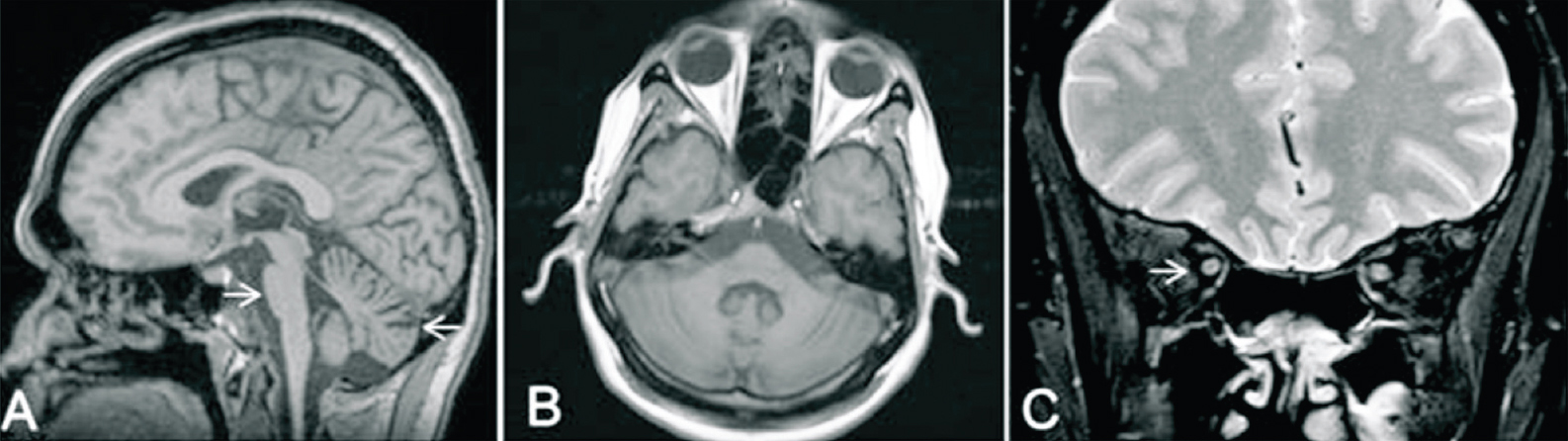
- Findings of MRI brain of a patient AT1319 (CAG)48 Saggital (A) and axial (B) reconstructed 3D-MPRAGE (magnetization-prepared rapid gradient-echo) images of brain showing flattening of anterior belly of pons suggesting pontine atrophy (shown by arrows) and prominent cerebellar folia suggest cerebellar atrophy. Coronal T2-W1 of orbit (C) shows no evidence of optic nerve atrophy (shown by arrows).
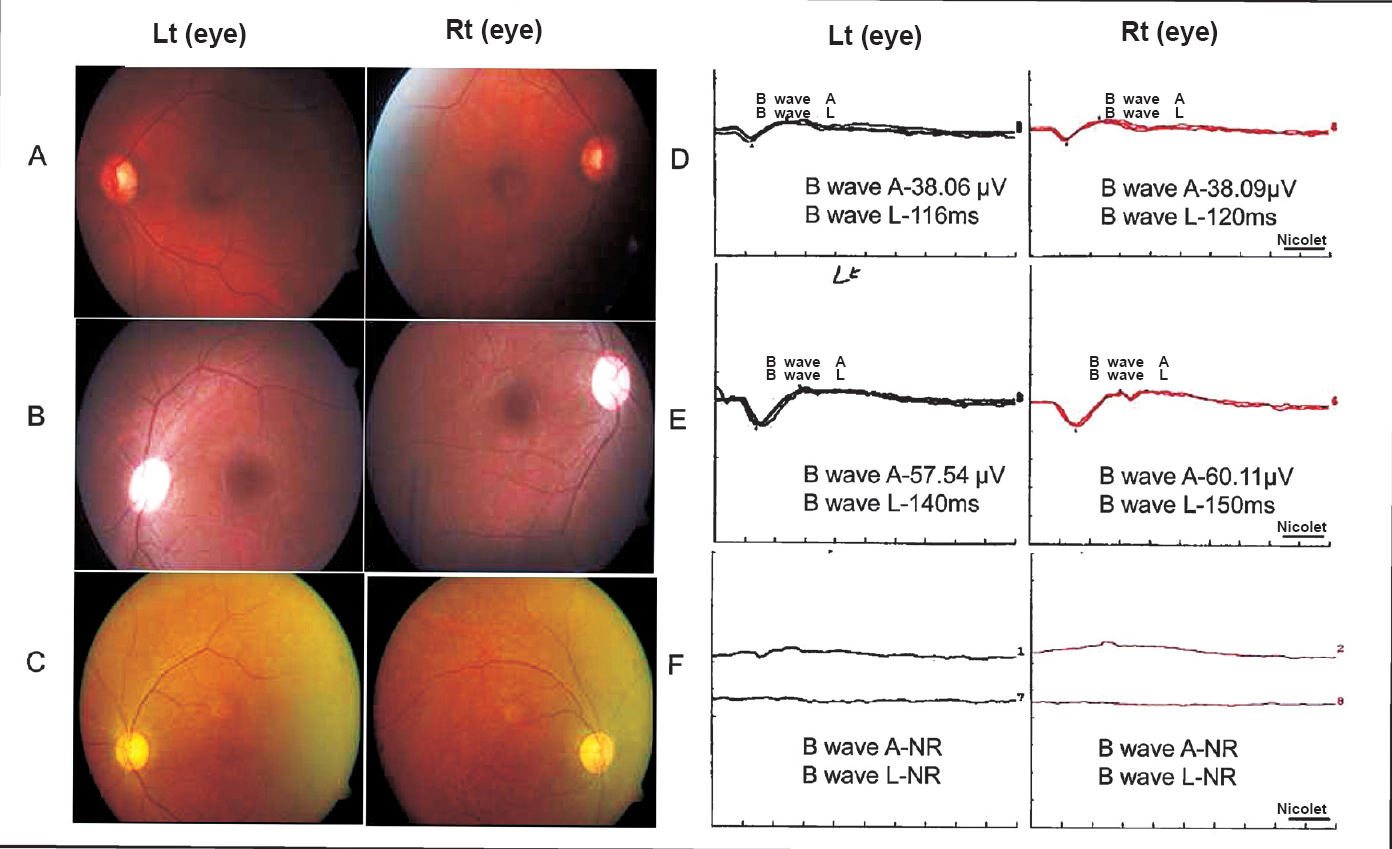
- Fundus images and electroretinography (ERG) tracing of representative patients. Fundus showing (A) AT1795 case (CAG)43; bilateral fine mottling of retinal pigmentary epithelium (RPE), (B) AT1839(CAG)54; essential normal appearing fundus both eyes, (C) AT236 (CAG)49; macular atrophy and mild retinal arterial attenuation. ERG recording- (D) AT1795; B wave amplitude (A, μV) reduction RE/LE (38.09/38.06), latencies (L, milliseconds) RE/LE (120/116), (E) AT1839; B wave amplitude normal in RE and mildly reduced in LE (57.54), latencies RE/LE (140/150), (F) AT236; Extinguished response in both eyes.
Clinico-genetic correlation: The Pearson's correlation analysis of repeat length and the age at onset showed a significant negative correlation (r= -0.73, P<0.01). We used quadratic regression model to fit the correlation between the age at onset (AO) and CAG repeat length and obtained the following standards: y[AO] = 0.0222[CAGn]2- 3.6084[CAG] + 148.19 (R2= 69.7). In 16 patients where cerebellar ataxia was scored for severity using SARA, mean (range) SARA score of 18.2(3-29) was observed. There was a positive correlation between CAG repeats adjusted SARA scores and the equation, y[CAG-adjusted-SARA-scores]=0.0234[Disease duration in years]+0.1798 (R2= 0.591) (Fig. 3.). The correlation of duration adjusted scores with repeat length was observed by equation, y[Duration-adjusted-SARA-scores] = 0.1867[CAG] - 6.4481(R2= 0.5495). Cerebellar ataxia preceded vision impairment in 10 patients with CAG repeat length 45.1±3.8 (range, 40-52). Only two patients had CAG repeat length 49 and 52, respectively and in the remaining cases it was <47. Higher CAG repeats 53.5±5.6 (range, 43-61) were observed in cases with vision deterioration preceding cerebellar ataxia or co-occurrence of the impairment. In the latter category most of the cases had CAG more than 48 except one patient with 43 CAG, vision and hand incoordination appeared simultaneously. The average duration to develop vision impairment (VI) in five patients, where gait ataxia preceded VI was 5.8 yr. This also was correlated inversely with the CAG repeat length (r= -0.83).
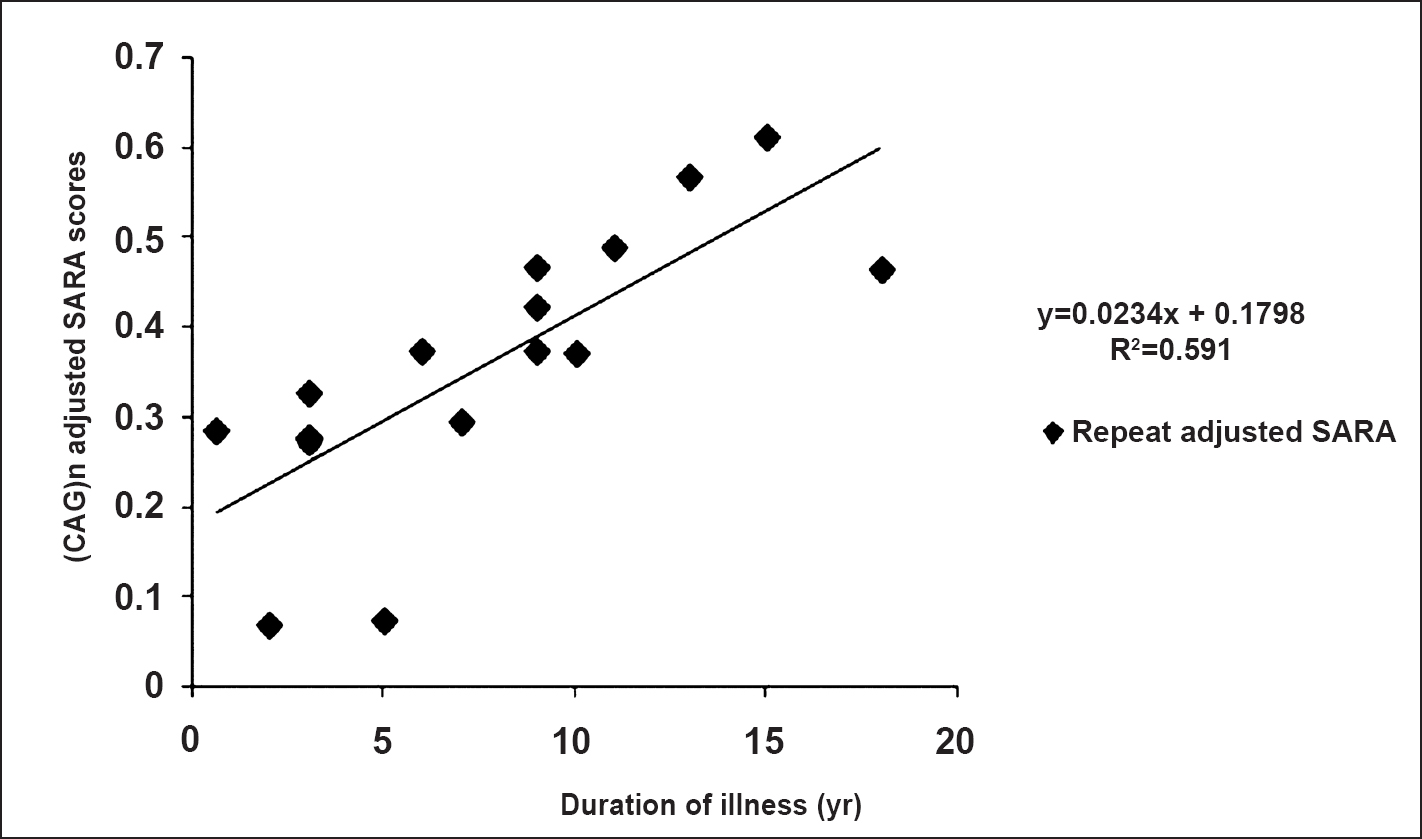
- Correlation of (CAG)n adjusted SARA scores with disease duration.
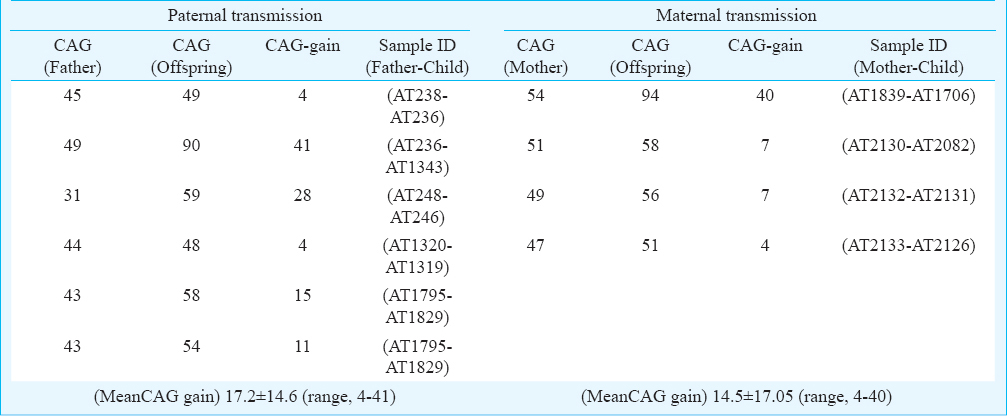
Intergenerational instability of CAG repeats: The average change in CAG repeats in 10 parent-child transmissions was 16.1 ± 14.7. Both the maternal-child and paternal-child transmission showed equal level of instability in the range of 4-40 and 4-41 CAG repeats, respectively (Table IV). One father-daughter transmission (AT2135→AT2136) was stable for 40 CAG repeats. No contraction of CAG repeat was observed during parental transmission of the repeats.
Pre-mutable normal CAG repeats were observed in a predisposed IE-N-LP2 population: The distribution of normal CAG repeat in 764 chromosomes of 21 Indian populations ranged from 9-34 with 10 CAG repeats being the most common allele observed occurring in 62 per cent of the total chromosomes studied. The larger distribution of CAG in the studied populations ranged from 9-16 except for IE-N-LP2 where three pre-mutable normal alleles of 28, 28 and 34 repeats were observed. The frequency estimation in terms of small normal (9-11) and large normal (>12) alleles is shown in Fig. 4. The cumulative frequencies of small and large normal CAG repeats were 0.74 and 0.26, respectively. Three populations unrelated ethnically to any of the SCA7 patients, IE-N-LP18, IE-W-LP4 and DR-S-LP915, had the higher frequencies of LN alleles compared to the other populations (SN/LN; 0.26/0.74, 0.5/0.5, 0.54/0.46).
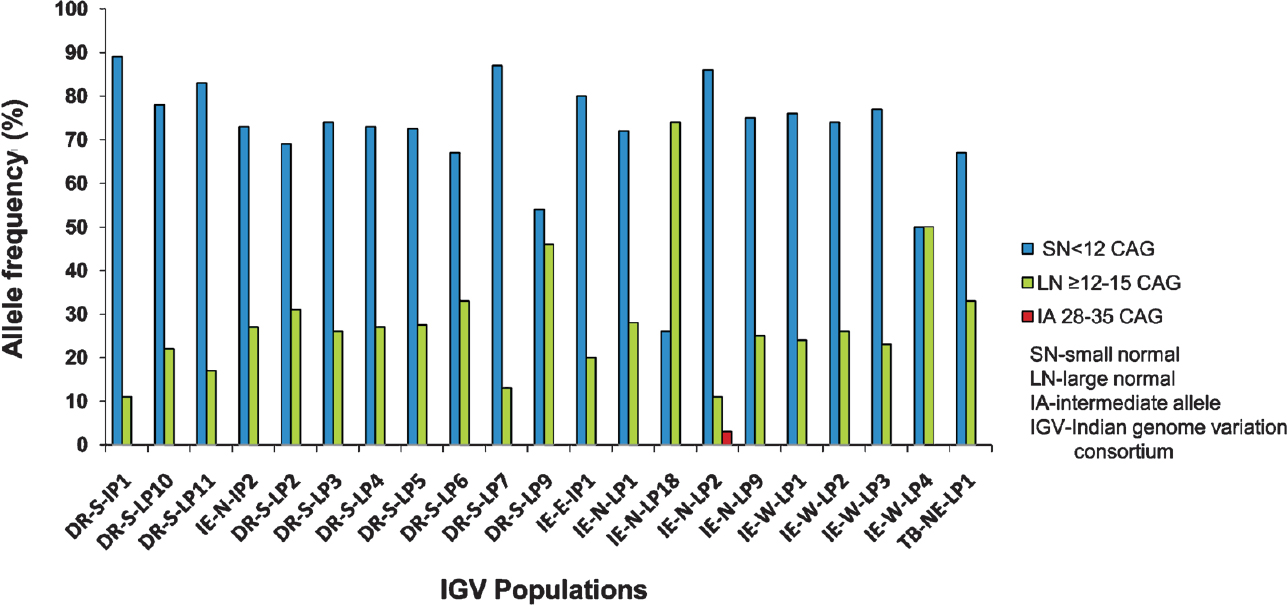
- CAG length distribution among healthy controls from 21 different ethnic populations of Indian origin.
Discussion
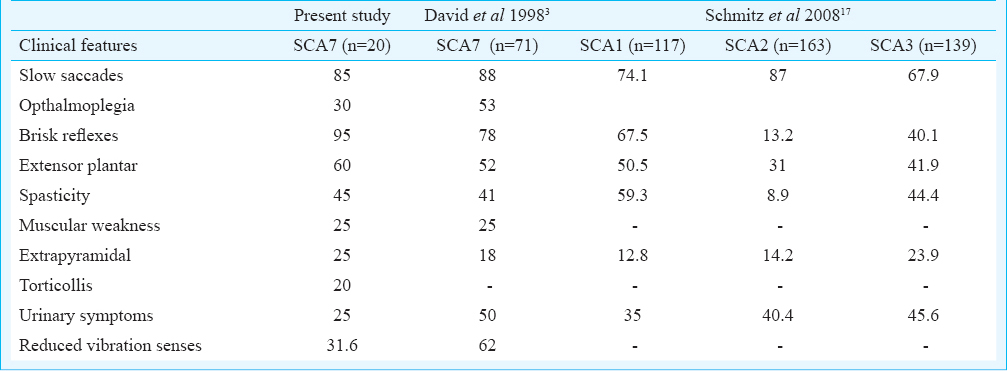
Our study provided the clinico-genetic analysis of nine Indian SCA7 families and CAG repeat distribution analysis in diverse Indian populations showed occurrence of ATXN7-CAG intermediate alleles in a predisposed population. The frequency of SCA7 in our study was 2.2 per cent. The clinical features observed in our patients confined to the essential phenotypic criteria of ADCA-II category129. As reported earlier, we also observed that slowing of saccadic velocity was prominent feature in our patients. David et al3 reported slow saccades in their 88 per cent (n=71) of SCA7 patients. Slowing of saccades is a characteristic oculomotor sign in SCA2; however, it is also observed in SCA1 (74%) and SCA3 (35-68%) but comparatively less in frequency171819. In both SCA2 and SCA7, slowing of saccades has been reported to appear early in the course of the disease1820. Neuropathologically and radiologically, both SCA2 and SCA7 share features of prominent pontine atrophy than cerebellum2122. The observed extracerebellar findings in our SCA7 patients were near similar to that observed in the large series of SCA7 patients by David et al3 except for reported frequency of sphincter disturbances was 50 per cent (25% in present study), reduced vibration senses 62 per cent (31.6% present study). This may be due to large sample size and disease duration in their study. The comparison of extracerebellar features in SCA7 (present data and David et al3) with other subtypes, SCA1-317 showed that SCA7 patients had combination of abnormalities generally over-represented in SCA1-3 types (Table III). Hyperreflexia and extensor plantar signs were observed to be more in SCA1, and in SCA7. This suggests a widespread involvement of brain region in SCA7 and comparatively more severe than other SCA types.
Duration of the disease and CAG repeats were observed to be determinant of SARA scores in our study, however, duration showed a better correlation with CAG-adjusted SARA scores. In a previous report23 disease severity was shown to correlate with CAG repeats, however, in their study disease severity was measured in terms of disease evolution (in decades) from detectable stage to debilitating stage. In SCA7, Ataxia→Vision impairment has been shown to be associated with low abnormal CAG length (47±6)3, (45±3.8 in present study) whereas a reverse symptom at onset pattern has been observed with high abnormal CAG length, >5910, 51±7310 and in the present study it was 53.5±5.6 CAG. This suggests early onset toxicity in the retinal tissue than in cerebellum from higher length of pathogenic polyglutamine stretch in ATXN7 protein. The trinucleotide repeat (TNR) expansions exhibit somatic instability which is reported to be tissue specific as shown in case of transgenic mice carrying ATXN7-CAG expansion and has also been observed in other TNR related genes e.g. Huntington disease2425. The somatic instability in case of ATXN7-CAG has been documented to be dependent on age as well as tissue as cortex, liver and kidney show more variability in transgenic mice model. It is speculated that a tissue specific dynamic change in methylation surrounding CAG may cause more instability in retinal tissue in CAG length dependent manner leading to early affection of vision. In one patient (AT1839) the fundus appeared normal, however, subtle abnormalities with amplitude reduction of B’ wave was observed in ERG. A previous report also suggests early subjective visual complaints and electrophysiological abnormalities in the absence of degenerative changes of fundus in SCA7 patients26. We also compared variation in age onset with categories of patients with CAG length <49 and >49 across reported SCA7 cases from different geographical region. (Table V). We observed that in earlier reported Chinese, Korean kindreds262728 and the present Indian study, SCA7 patients had comparatively younger mean age at onset 30.8, 36 and 33.4yr,respectively in CAG<49 category. The study from Sweden10 reported 40.5yr and data from mixed ethnicity study reported 40 yr3 being mean age at onset in CAG<49 group (Table V). We observed moderate degree of association of younger onset with<49 CAG repeats in Asian SCA7 patients which indicated population specific non-CAG genetic or other unknown factors contributing to early degeneration of cerebellum and associated regions.

The analysis of inter-generational instability of repeats revealed one de novo expansion from mutable intermediate repeat 31 to pathological size 59 which has been reported earlier by our group14, in addition, one maternal and one paternal transmission led to gain in repeat size of +41 and 40, respectively. Benton et al29 observed a highly unstable maternal transmission in the range of -1 to +56. This was in contrary to earlier reports of higher degree of instability. Gouw et al23 reported mean gain of repeats of 16.6±11 (range -6 to +62) during paternal and 3.8±3 (range -13 to 17) by maternal transmission in the offspring generation, while David et al3 reported higher paternal instability of 15 ± 20(range 0 to +85) than the maternal +5 ± 5(range -6 to +14) transmission. Overall, these findings suggest that both male and female gametogenic events can contribute to the instability of the CAG stretch of ATXN7.
The analysis of CAG repeat length distribution among diverse Indian populations allowed us to identify intermediate length CAG alleles at SCA7 locus (ATXN7) in IE-N-LP2 population. We have not identified CAG repeats length beyond 16 in any other Indian population (Fig 4). The frequency of large normal alleles in IE-N-LP2 was comparatively low (11%) than in other populations. The commonest repeat length observed among controls was 10 repeats in all the subgroup populations except for IE-N-LP18 where 12-CAG repeats containing ATXN7 being observed in higher frequency and so were the large normal alleles (0.74). The other two populations IE-W-LP4 and DR-S-LP9 also showed higher frequency for LN alleles (0.5, 0.46). In case of SCA7, we observed three intermediate alleles 28-34 and there was an allelic gap of 17-27 CAG repeats among healthy control population. We hypothesize that origin of SCA7 can largely be governed by formation of 28 CAG allele containing chromosome from normal alleles during any of the DNA synthesis or repair events such as, replication, gene conversion, recombination, duplication or some yet unknown mechanism. Alternatively, it is also possible that alleles in range of 20-27 are lethal and are negatively selected from the population.
In conclusion, SCA7 in India seems to be low though there may be independent events of recurrent generation of IA in different populations. A higher degree of CAG instability upon transmission across the generation results in change in repeat length and that results in anticipation phenomenon. The phenotypic homogeneity is observed worldwide but variable phenotypic expressions also exist. From ophthalmologic point of view ERG may serve as an early para-clinical investigation for at risk individuals of SCA7 families for diagnosing retinal impairment. The detailed haplotype and sequence analysis of ATXN7 in these families and intermediate alleles would be warranted to identify genetic mechanism implicated in recurrent generation of IA alleles.
Acknowledgment
This study was carried out through CSIR (Council of Scientific and Industrial Research) funded project SIP0006. Authors thank all the participating members from the affected families for their regular cooperation in clinical and genetic investigations. Help received from Dr Inder Singh (Neurology Department, AIIMS) in blood collection, Shri Akhilesh Kumar Sonkar (CSIR-IGIB, Neurology Department, AIIMS) in patient calling, follow up and for providing help in obtaining various investigational records for the participating affected individuals from various institutional departments is duly acknowledged.
References
- Autosomal dominant cerebellar ataxia with retinal degeneration: clinical, neuropathologic, and genetic analysis of a large kindred. Neurology. 1994;44:1441-7.
- [Google Scholar]
- Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet. 1997;17:65-70.
- [Google Scholar]
- Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7) Hum Mol Genet. 1998;7:165-70.
- [Google Scholar]
- Molecular genetic analysis of autosomal dominant cerebellar ataxia with retinal degeneration (ADCA type II) caused by CAG triplet repeat expansion. Hum Mol Genet. 1998;7:177-86.
- [Google Scholar]
- Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet. 2004;12:2-15.
- [Google Scholar]
- Evidence for a common Spinocerebellar ataxia type 7 (SCA7) founder mutation in Scandinavia. Eur J Hum Genet. 2000;8:918-22.
- [Google Scholar]
- Autosomal dominant cerebellar ataxia with pigmentary macular dystrophy. A clinical and genetic study of eight families. Brain. 1994;117:445-60.
- [Google Scholar]
- Molecular and clinical study of 18 families with ADCA type II: evidence for genetic heterogeneity and de novo mutation. Am J Hum Genet. 1999;64:1594-603.
- [Google Scholar]
- Expanded CAG repeats in Swedish spinocerebellar ataxia type 7 (SCA7) patients: effect of CAG repeat length on the clinical manifestation. Hum Mol Genet. 1998;7:171-6.
- [Google Scholar]
- Multiple origins of the spinocerebellar ataxia 7 (SCA7) mutation revealed by linkage disequilibrium studies with closely flanking markers, including an intragenic polymorphism (G3145TG/A3145TG) Eur J Hum Genet. 1999;7:889-96.
- [Google Scholar]
- SCA-LSVD: a repeat-oriented locus-specific variation database for genotype to phenotype correlations in spinocerebellar ataxias. Hum Mutat. 2009;30:1037-42.
- [Google Scholar]
- Molecular analysis of CAG repeats at five different spinocerebellar ataxia loci: correlation and alternative explanations for disease pathogenesis. Mol Cells. 2007;24:338-42.
- [Google Scholar]
- Post-zygotic de novo trinucleotide repeat expansion at spinocerebellar ataxia type 7 locus: evidence from an Indian family. J Hum Genet. 2005;50:155-7.
- [Google Scholar]
- Indian Genome Variation Cosortium. Genetic landscape of the people of India: a canvas for disease gene exploration. J Genet. 2008;87:3-20.
- [Google Scholar]
- Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66:1717-20.
- [Google Scholar]
- Spinocerebellar ataxia types 1, 2, 3, and 6: disease severity and nonataxia symptoms. Neurology. 2008;71:982-9.
- [Google Scholar]
- A clinicogenetic analysis of six Indian spinocerebellar ataxia (SCA2) pedigrees. The significance of slow saccades in diagnosis. Brain. 1998;121:2341-55.
- [Google Scholar]
- Autosomal dominant cerebellar ataxia type I: oculomotor abnormalities in families with SCA1, SCA2, and SCA3. J Neurol. 1999;246:789-97.
- [Google Scholar]
- Slowing of voluntary and involuntary saccades: an early sign in spinocerebellar ataxia type 7. Ann Neurol. 2001;49:801-4.
- [Google Scholar]
- Pontine atrophy precedes cerebellar degeneration in spinocerebellar ataxia 7: MRI-based volumetric analysis. J Neurol Neurosurg Psychiatry. 2004;75:1452-6.
- [Google Scholar]
- Spinocerebellar ataxia type 7 (SCA7): widespread brain damage in an adult-onset patient with progressive visual impairments in comparison with an adult-onset patient without visual impairments. Neuropathol Appl Neurobiol. 2008;34:155-68.
- [Google Scholar]
- Analysis of the dynamic mutation in the SCA7 gene shows marked parental effects on CAG repeat transmission. Hum Mol Genet. 1998;7:525-32.
- [Google Scholar]
- Quantification of age-dependent somatic CAG repeat instability in Hdh CAG knock-in mice reveals different expansion dynamics in striatum and liver. PLoS One. 2011;6:e23647.
- [Google Scholar]
- CTCF cis-regulates trinucleotide repeat instability in an epigenetic manner: a novel basis for mutational hot spot determination. PLoS Genet. 2008;4:e1000257.
- [Google Scholar]
- Trinucleotide expansions in the SCA7 gene in a large family with spinocerebellar ataxia and craniocervical dystonia. Neurosci Lett. 2008;434:230-3.
- [Google Scholar]
- Molecular and clinical study of spinocerebellar ataxia type 7 in Chinese kindreds. Arch Neurol. 2000;57:1513-8.
- [Google Scholar]
- Clinical and neuroradiological features of patients with spinocerebellar ataxias from Korean kindreds. Arch Neurol. 2003;60:1566-74.
- [Google Scholar]
- Molecular and clinical studies in SCA-7 define a broad clinical spectrum and the infantile phenotype. Neurology. 1998;51:1081-6.
- [Google Scholar]




