
Translate this page into:
Rapid molecular diagnosis of spinal muscular atrophy
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Spinal muscular atrophy (SMA) is a group of inherited autosomal neuromuscular disorder which is characterized by progressive muscle weakness and is the leading genetic cause of childhood deaths. The SMA is clinically heterogenous and clinical spectrum ranges from early infant death to normal adult life with only mild weakness. The International SMA Consortium has subdivided the disease into three types according to age at first symptoms and milestones of development. Type I SMA is the severe form of Werdnig-Hoffmann disease with onset at birth or before 6 months and death due to respiratory distress usually within 2 years. Actually, type I SMA patients are never able to sit or walk due to profound muscular weakness. Children with type II SMA (intermediate form) can sit but cannot stand or walk unaided. In type III SMA (Kugelberg- Welander disease) patients show the first clinical signs after 18 months, evolving to a chronic course1.
The most common form of SMA is caused by mutations of the spinal motor neuron (SMN) gene. The identification of the SMN as the SMA-causing gene by Lefebvre et al2 has been an important milestone in the understanding of the molecular basis of this devastating neuromuscular disorder. This gene is part of 500 kb inverted duplication on chromosome 5q13 and the duplicated region contains at least four genes and repetitive elements which make it prone to rearrangements and deletions3.
There are at least two SMN genes, termed as telomeric (SMNt) and other centomeric (SMNc) on each chromosome. The telomeric and centromeric copies of SMN genes are nearly identical and encode the same protein. However, mutations in telomeric copy of the gene are associated with spinal muscular atrophy; whereas mutations in the centromeric copy do not lead to SMA. The centromeric gene may result from mutation in the telomeric copy and believed to be a modifier of disease. Although several nucleotides are different in telemeric and centromeric genes (Fig. 1) but the critical sequence difference between the two genes is a single nucleotide in exon 7 which is thought to be an exon splice enhancer. Earlier, the two genes were referred to as SMNt and SMNc but now these have been redesignated as SMN1 and SMN2, respectively.
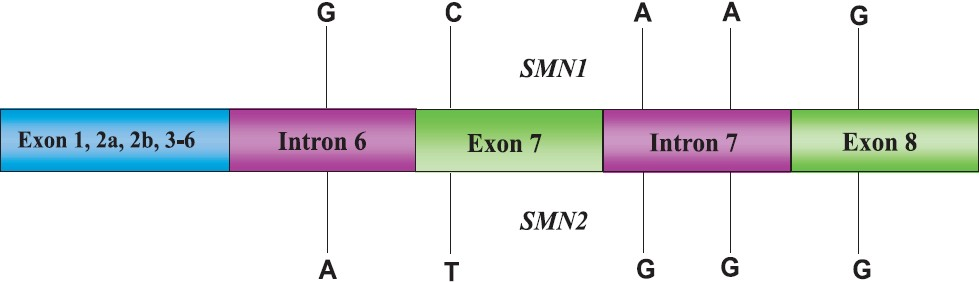
- Nucleotide differences in telomeric (SMN1) and centromeric (SMN2) copies of SMN genes.
For DNA based mutation detection, one bp variation each in exons 7 and 8 between SMN1 and SMN2 forms the basis of detecting disease gene in the patients. The conventional method of differentiation is PCR-RFLP in which same size product is obtained from both SMN1 and SMN2. However, after restriction digestion, only products of exon 7 and exon 8 of SMN2 are cleaved by restriction enzymes Dra1 and Dde1, respectively and presence/absence of the two genes can be determined after gel electrophoresis. Both the restriction enzymes are relatively expensive and incomplete digestion can sometime pose a problem in the correct diagnosis. In recent years, a modification of PCR termed as allele-specific (AS)-PCR also known as amplification refractory mutation system (ARMS) has been increasingly utilized to differentiate single base changes by PCR itself, thus avoiding restriction digestion. Initially, allele-specific PCR was carried out by using two different forward primers, each ending with specific nucleotide at 3’ end while a common reverse primer was used. These reactions were carried out in two tubes and primers matching particular allele could only give the product. Although the principle and technology were straight forward but there were mis-diagnosis particularly for heterozygotes. Therefore, further refinement of technology has taken place and two pairs of primers are used to amplify two alleles in single tube PCR reaction. The primers are designed so that the two primer pairs overlap at a SNP location but each match perfectly to only one of the possible SNPs (Fig. 2). If a given allele is present in the PCR reaction, the primer pair specific to that allele will yield product but not to the alternative allele with a different SNP. The two primer pairs are also designed keeping in mind that their PCR products are significantly different in length, thus allowing for easily distinguishable bands after electrophoresis. The samples containing homozygous alleles will show two types of PCR products, one that results from the inner primer (which matches the SNP location) and the outer primer and the other that results from the two outer primer pairs. If the genomic sample is heterozygous, then three types of PCR products will be formed, two from the inner primer of each allele to their respective outer primer pairs and other from the two opposite, outer primers (Fig. 2). The critical step is the designing of appropriate primers and web-based tools are also available4. Of course, the designed primers need to be tested in the known samples to ensure correct genotyping.
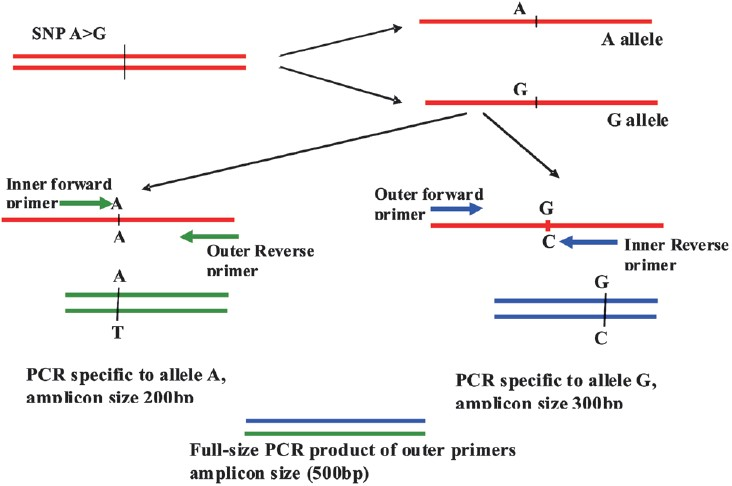
- Diagrammatic representation of allele specific PCR using tetraprimers: In the above hypothetical example, full amplicon size of 500 bp is considered. The primers here have been designed in such a way that the presence of allele A will give a band of 200 bp in the PCR amplification whereas 300 bp size band will be seen for G allele. Simultaneously, a common PCR product of two outer primers (500 bp) will also be obtained. After gel electrophoresis, all three PCR products can be resolved. Homozygous A genotypes will show two PCR bands of 200 and 500 bp, homozygous G genotypes will correspond to 300 and 500 bp, while presence of three bands (200, 300 and 500 bp) will indicate heterozygotes (A/G).
For SMA diagnosis, Baris et al5 recently used tetraprimer approach to distinguish SMN1 and SMN2 and claimed their diagnosis to completely match with PCR-RFLP. However, in molecular pathology laboratories, varying amounts of DNA are used, as only semi-quantitative estimation is carried out. Using too much or too little DNA can be problematic for accurate determination. In the accompanying article in this issue, Marini et al6 have used only a single primer pair which only amplifies SMN1, not SMN2. In order to ensure that absence of PCR band is not due to failure of PCR reaction, they have added second set of primers belonging to β-globin gene. To keep the assay simplified, they have looked for the absence of only exon 7 as the causative mutation in SMA. Actually, it was recently reported by the same group7 that detection of exon 7 mutation alone is sufficient for correct diagnosis of SMA. Therefore, any pathological laboratory having thermocycler and gel electrophoresis equipment can provide molecular diagnosis of SMA in 1-2 days and test is also cost-effective. The rapidity of this method ensures a much shorter turn around time of sample processing which is more appealing to referring clinicians.
Once the mutation is confirmed, the family can be referred for genetic counselling and a prenatal test can be offered in next pregnancy. It may be added that although absence of SMN1 gene is a confirmatory test but age and clinical presentation are also important considerations for predicting future course of the disease. The copy number of SMN2 is also known to affect the clinical presentation but the quantitative SMN2 copy number test requires expensive equipment like real-time PCR and careful standardizations. Therefore, SMN2 copy number test is not routinely carried out in the diagnostic laboratories. A negative SMN result does not completely rule out SMA as approximately 5 per cent of patients have point mutations instead of gross deletion of SMN1 gene, and these point mutations also cannot be detected by the PCR-based techniques discussed here6. There is no cure for SMA although various novel experimental approaches are being tried in animal models8.
Availability of a reliable simplified test is one aspect of clinical diagnosis of genetic diseases but carrier detection is equally important for prevention of the disease. Although, claims have been made for correct carrier determination and SMN2 dosage analysis but the tests are still technically challenging. As the availability and usage of real-time PCR are becoming more common, it should be possible to develop simple, economical and reliable tests for mutation analysis based on newer technologies like high-resolution melting curve analysis.
References
- Spinal muscular atrophy: a clinical and research update. Pediatr Neurol. 2012;46:1-12.
- [Google Scholar]
- Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155-65.
- [Google Scholar]
- Clinical and molecular diagnosis of spinal muscular atrophy. Neurol India. 2002;50:117-22.
- [Google Scholar]
- WASP: a Web-based Allele-specific PCR assay designing tool for detecting SNPs and mutations. BMC Genomics. 2007;8:275.
- [Google Scholar]
- Rapid diagnosis of spinal muscular atrophy using tetra-primer ARMS PCR assay: simultaneous detection of SMN1 and SMN2 deletion. Mol Cell Probes. 2010;24:138-41.
- [Google Scholar]
- Allele-specific PCR for a cost-effective & time-efficient diagnostic screening of spinal muscular atrophy. Indian J Med Res. 2012;135:31-5.
- [Google Scholar]
- Deletion analysis of SMN1 exon 7 alone may be necessary and sufficient for the diagnosis of spinal muscular atrophy. J Neurogenet. 2011;25:15-6.
- [Google Scholar]
- New therapeutic approaches to spinal muscular atrophy. Curr Neurol Neurosci Rep 2011 In press
- [Google Scholar]




