
Translate this page into:
Possibility of re-purposing antifungal drugs posaconazole & isavuconazole against promastigote form of Leishmania major
For correspondence: Dr Rakesh Sehgal, Aarupadai Veedu Medical College & Hospital, Kirumampakkam, Puducherry 607 402, India e-mail: sehgalpgi@gmail.com
-
Received: ,
Accepted: ,
Abstract
Background & objectives
The emergence of drug resistance in leishmaniasis has remained a concern. Even new drugs have been found to be less effective within a few years of their use. Coupled with their related side effects and cost-effectiveness, this has prompted the search for alternative therapeutic options. In this study, the Computer Aided Drug Design (CADD) approach was used to repurpose already existing drugs against Leishmania major. The enzyme lanosterol 14-alpha demethylase (CYP51), in L. major, was chosen as the drug target since it is a key enzyme involved in synthesizing ergosterol, a crucial component of the cell membrane.
Methods
A library of 1615 FDA-approved drugs was virtually screened and docked with modeled CYP51 at its predicted binding site. The drugs with high scores and high affinity were subjected to Molecular Dynamics (MD) simulations for 100 ns. Finally, the compounds were tested in vitro using an MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide] assay against the promastigotes of L. major.
Results
Computational screening of FDA-approved drugs identified posaconazole and isavuconazole as promising candidates, as both drugs target the CYP51 enzyme in fungi. Molecular dynamics (MD) simulations demonstrated that both drugs form stable complexes with the target enzyme. In vitro studies of posaconazole and isavuconazole against promastigotes of L. major demonstrated significant efficacy, with IC50 values of 2.062±0.89 µg/ml and 1.202±0.47 µg/ml, respectively.
Interpretation & conclusions
The study showed that the existing FDA-approved drugs posaconazole and isavuconazole can successfully be repurposed for treating L. major by targeting the CYP51 enzyme, demonstrating significant efficacy against promastigotes.
Keywords
CADD
isavuconazole
Leishmania major
molecular dynamics (MD) simulation
MTT assay
posaconazole
virtual screening

Leishmaniasis is a disease caused by protozoan parasites from more than 20 different Leishmania species. It presents in three primary forms: visceral leishmaniasis (VL), cutaneous leishmaniasis (CL), and mucocutaneous leishmaniasis1,2. VL, the most severe form, is fatal if untreated in over 95 per cent of cases and is characterized by symptoms like fever, weight loss, and organ enlargement. It primarily affects regions such as Brazil, East Africa, and India3,4. CL causes skin ulcers and is prevalent in the Americas, the Mediterranean basin, the Middle East, and Central Asia. Mucocutaneous leishmaniasis leads to the destruction of mucous membranes in the nose, mouth, and throat, with most cases found in Bolivia, Brazil, Ethiopia, and Peru5,6. Leishmaniasis disproportionately affects the world’s poorest populations, associated with factors like malnutrition, poor housing, and weakened immune systems. Despite an estimated 700,000 to 1 million new cases annually, only a small fraction develops symptoms. The disease's epidemiology varies by region, with different forms and species prevalence across Africa, the Americas, the Eastern Mediterranean, Europe, and South-East Asia7.
Leishmania has developed resistance to key drugs like antimony, miltefosine, and amphotericin B, complicating treatment efforts. Antimony resistance in Leishmania is primarily due to reduced drug uptake facilitated by the downregulation of aquaglyceroporin 1 (AQP1) and increased intracellular thiol levels, which enhance the parasite's antioxidant capacity. Additionally, increased drug efflux through ATP-binding cassette (ABC) transporters and modulation of host immune responses contribute to resistance8,9. Miltefosine resistance involves decreased drug accumulation, overexpression of multidrug resistance proteins like MRPA, and genetic mutations affecting lipid metabolism and transporter activity. These mechanisms are often stable and specific to miltefosine, with some strains also showing cross-resistance to other drugs10-12. Amphotericin B (AmB) resistance is associated with mutations in the sterol biosynthesis pathway, leading to altered membrane sterol composition and decreased drug binding affinity. This resistance can result from both laboratory-induced mutations and natural selection in clinical settings13-15. The complexity and persistence of these resistance mechanisms highlight the urgent need for new therapeutic approaches, including novel drugs and drug combinations, to effectively treat resistant strains of Leishmania.
A lot of drug targets have been explored in leishmania to seek new therapeutic opportunities. Squalene epoxidase, 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGR) enzyme from sterol biosynthetic pathway16,17, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and phosphoglycerate kinase from glycolytic pathway18, dihydrofolate reductase (DHFR) and pteridine reductase 1(PTR1) from folate biosynthesis pathway19,20, trypanthione reductase from trypanothione pathway21, spermidine synthase from hypusine pathway22, L-asparaginase23 etc., have been extensively studied as their potency as drug targets. Lanosterol 14-alpha demethylase from the sterol biosynthetic pathway in Leishmania is a promising candidate due to its crucial role in sterol biosynthesis, which is essential for maintaining the parasite's cell membrane integrity. Lanosterol 14-alpha demethylase (CYP51) is an attractive drug target in Leishmania as it has a unique role in ergosterol biosynthesis, a pathway absent in humans that minimizes off-target effects and enhances safety. This specificity allows for a broader therapeutic window, potentially enabling higher doses without host toxicity. The extensive research on CYP51 in fungi provides valuable insights into drug development, resistance mechanisms, and pharmacokinetics, facilitating the design of effective inhibitors. Additionally, the fitness cost associated with resistance mutations in CYP51 may limit the spread of resistant strains, offering a significant advantage over other targets where resistance can develop more readily24,25.
Repurposing the existing drugs stands out as a viable solution due to its efficiency in terms of time, ease, and cost-effectiveness. Hence, this research focuses on repurposing FDA-approved drugs against L. major through virtual screening and molecular docking. Molecular Dynamics (MD) simulation studies were conducted to study the behaviour and stability of the selected drugs with CYP51 over a span of 100 ns. Finally, an in vitro test was conducted to assess the effectiveness of the selected drugs against the survival of the promastigote form of the parasite.
Materials & Methods
The study was conducted at the department of Medical Parasitology, Post Graduate Institute of Medical Education and Research, Chandigarh, Punjab, India from December 2021 to January 2023.
Structural preparation of protein
The amino acid sequence of CYP51 protein from Leishmania major, strain Friedlin was retrieved from the NCBI Database26. Since the three-dimensional structure of this protein was not available in the Protein Data Bank27 or UniProt28, a homology model was generated using the SWISS Model server29. To ensure the accuracy of the modeled structure, validation was conducted using the Structural Analysis and Verification Server (SAVES) version 6.0 ( https://saves.mbi.ucla.edu/ ). Using SAVES, ERRAT ( https://www.doe-mbi.ucla.edu/errat/ ) was employed to evaluate the quality of the structure, and PROCHECK ( https://www.ebi.ac.uk/thornton-srv/software/PROCHECK/ ) was used to plot the Ramachandran plot of the modeled protein30. After the structural analysis, the protein was subjected to preprocessing using Discovery Studio ( https://www.3ds.com/products/biovia/discovery-studio ) to optimize the protein structure for precise virtual screening and docking experiments. This involved fixing misoriented groups, creating missing disulfide bonds, removing heteroatoms, deleting water molecules, and adding hydrogen atoms31.
Prediction of the binding site of proteins
To predict the binding site of the modelled protein, multiple computational tools were employed, including Prankweb32, ProBIS33, COACH34, and Discovery Studio31. Each of these tools provided a distinct method for identifying potential binding sites on the protein's surface, allowing for a thorough and comprehensive analysis. After running the protein through each software, the binding sites were individually identified and carefully compared. From this comparative analysis, a consensus sequence was derived, representing the common binding sites predicted by all four tools. This consensus sequence was then utilized to define and generate a grid box, which was essential for conducting accurate virtual screening and molecular docking studies.
Creation of drug library and preparation of the ligands
A drug library containing 1,615 FDA-approved drugs was compiled from the ZINC database35. Each drug was validated for FDA approval, and duplicates or invalid entries were removed. The library was then preprocessed using Open Babel, which involved converting molecular formats, generating multiple conformations, assigning charges, and adding hydrogen atoms to the ligands36. These steps ensured that the drug library was optimized for accurate virtual screening and molecular docking experiments.
Virtual screening and molecular docking
PyRx 0.8 software ( https://pyrx.sourceforge.io/ ) was used to conduct virtual screening and molecular docking, an effective method for identifying potential drug compounds during the drug discovery process37. The drug library, derived from the ZINC database, was initially in .sdf format and contained all the compounds in a single file. To prepare the file for analysis in PyRx 0.8, it was imported from Open Babel. Afterwards, the energy of the ligands was minimized, and all the ligands were converted into AutoDock PDBQT format for further analysis.
On the other hand, the modeled protein was imported into PyRx 0.8. Its energy was minimized, and the protein was converted into PDBQT format. Subsequently, a grid box was generated by using the binding sequence of CYP51. The dimensions of the grid box were as follows: center (X,Y,Z: 32.50, -27.004, 0.5782) and size (X,Y,Z: 29.089, 28.917, 30.276) with an exhaustiveness of 8. Using PyRx 0.8 and AutoDock Vina, CYP51 was docked within the grid box along with the drug library to study their interactions. The ligands that showed the highest binding energy score and formed a significant number of hydrogen bonds with CYP51 were selected for further analysis. The selected complex docking result was further validated through Autodock 438.
Molecular dynamics (MD) simulation and trajectory analysis
The selected protein-ligand complexes from virtual screening were analyzed for molecular dynamic simulation to evaluate their stability. The stability assessment was performed using the Desmond v5.6 module of Schrödinger-Maestro v11.8 in a Linux environment39. The simulations were conducted for a period of 100 nanoseconds, employing the Optimized Potentials for Liquid Simulations (OPLS) force field. The simulations were executed at a temperature of 300 Kelvin, following normal pressure and temperature (NPT) conditions.
Trajectory analysis of the molecular dynamics (MD) simulation was used to evaluate structural deviation using Root Mean Square Deviation (RMSD), atomic fluctuations with Root Mean Square Fluctuation (RMSF), and protein-ligand interactions through Protein-Ligand (PL) contacts39,40.
Antileishmanial Potential of posaconazole and isavuconazole against promastigote
The promastigotes of MHOM/SU/73/5ASKH strain of Leishmania major were cultured in RPMI-1640 medium supplemented with 10 per cent FBS and antibiotics (gentamycin and streptomycin) at an ambient temperature (22±1° C) in a biochemical oxygen demand (BOD) incubator. The growing culture was sub-cultured every 48-72h in fresh medium. The effectiveness of posaconazole and isavuconazole against the promastigote stage of L. major was assessed using the MTT cell viability assay41. Promastigotes, with a cell density of 2 × 105 cells/ml, were cultured with varying concentrations (ranging from 0 to 50 μg/ml) of the test drugs in a BOD incubator for 72h at 24°C. This incubation took place in a 96-well tissue culture plate. Paromomycin was used as a control. The inhibitory concentration of the test drugs (IC50) was determined through extrapolation on a graph (using GraphPad Prism 9.0). The experiments were performed in triplicates and repeated three times.
Results
Modeling and validation of 3D structure of CYP51
The PSI-BLAST algorithm utilized by the Swiss Model was employed to model the 3D structures of CYP51 using the closest homologous protein structures as templates (Fig. 1; panel A). GMQE value on a scale of 0 to 1, served as an indicator of the quality of modeled structure, with scores exceeding 0.8 suggesting high quality (Table I). Over 90 per cent of residues in all modeled structures resided in the most favored regions of the Ramachandran plot, affirming overall high quality (Fig. 1B). Additionally, ERRAT values, assessing protein structure quality via electron density agreement, were above 90, indicating high quality (Fig. 1C). The modeled PDB structure was then imported into Discovery Studio for preprocessing, following the outlined methodology and then saved in .pdb format.
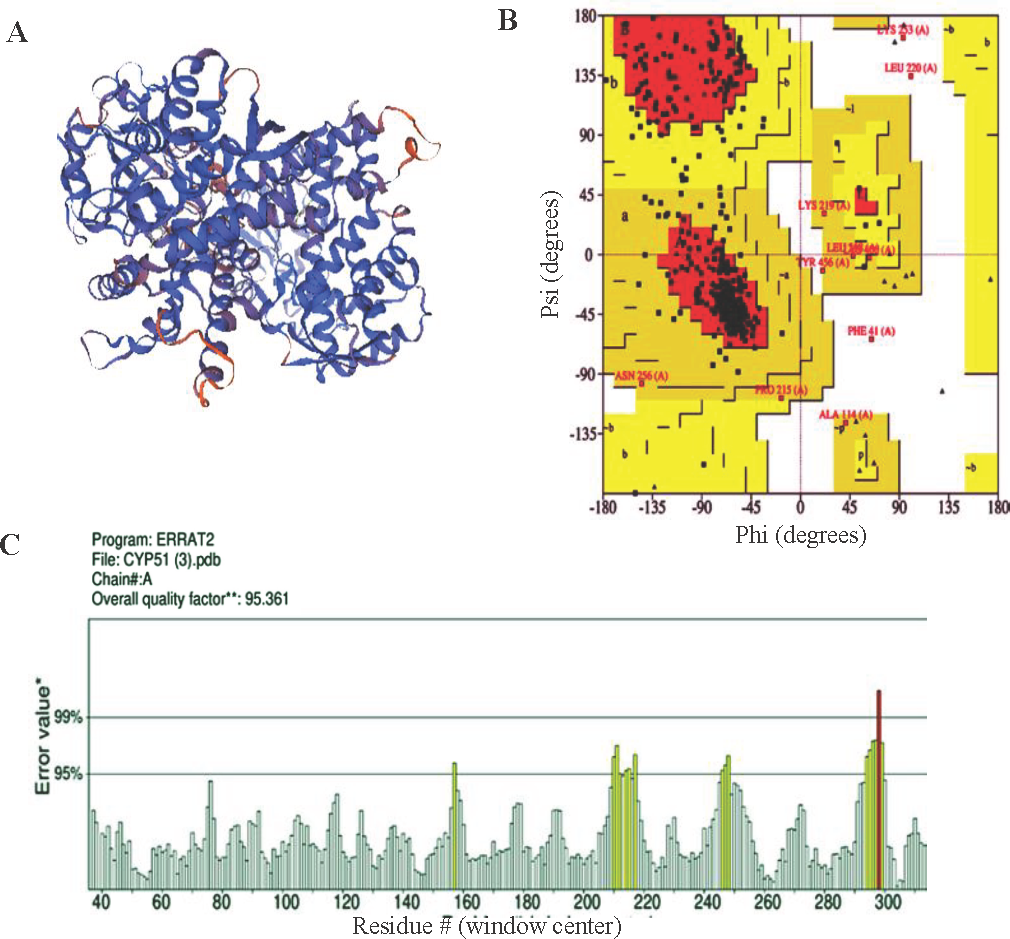
- CYP51 (A) 3D model, (B) Ramachandran plot and (C) ERRAT value.
| Putative drug target | Template | GMQE | ERRAT value | Residues in most favored region (Ramachandran plot) |
|---|---|---|---|---|
| CYP51 | 3l4d.1.A | 0.85 | 95.10 | 94% |
Binding sites of the protein
The binding sites of the CYP51 protein were identified using Prankweb, ProBIS, COACH, and Discovery Studio. Each tool provided unique predictions based on different algorithms, such as deep learning, structural comparison, and integration of multiple scoring functions. The results from these tools were then compared, and a consensus sequence of binding sites was derived from overlapping predictions (Table II). This consensus sequence was used to define the grid box for molecular docking, ensuring that all potential binding regions were accurately targeted in subsequent studies.
| Tools | Binding site sequences |
|---|---|
| PrankWeb | 45, 48,49, 52, 56, 69, 71, 76, 89, 102, 104, 105, 109, 114, 115, 126, 123, 133, 178, 182, 209, 210, 213, 226, 233, 283, 286, 287, 288, 289, 290, 291, 292, 294, 295, 298, 349, 354, 355, 356, 357, 358, 359, 360, 415, 416, 417, 420, 421, 422, 423, 424, 427, 428, 432, 456, 458, 459, 460 |
| ProBIS | 102, 115, 123, 126, 129, 133, 287, 290, 291, 294, 349, 354, 355, 358, 360, 415, 420, 421, 422, 423, 427 |
| COACH | 9, 114, 122, 126, 133, 287, 290, 291, 294, 295, 298,, 349, 354, 355, 358, 360, 383, 414, 415, 416, 417, 420, 422, 423, 424, 428 |
| Discovery studio | 102, 115, 123, 126, 129, 133, 287, 290, 291, 294, 349, 354, 355, 358, 360, 415, 420, 421, 422, 423, 427 |
| Consensus sequence | 126, 133, 287, 291, 294, 349, 354, 355, 358, 360, 415, 420, 422, 423 |
Virtual screening and molecular docking of CYP51
Virtual screening and molecular docking analysis were conducted on CYP51 using a library of FDA-approved drugs. The docking results were evaluated based on binding energies and the number of interactions, particularly hydrogen bonds, with key residues of CYP51, with fluconazole used as a target-specific control compound due to its established role in inhibiting CYP51 in fungi (Table III). A threshold was set for binding energies at or below -9 kcal/mol and a minimum of three hydrogen bonds. Posaconazole (Fig. 2) and isavuconazole (Fig. 3) were identified as top candidates due to their strong binding affinities and similar mechanisms of action targeting CYP51 in fungi, making them promising for further study. The docking results of the selected drug-protein complexes were further validated using AutoDock4, where the binding free energies of posaconazole, isavuconazole and fluconazole were found to be -3.54 kcal/mol, -6.03 kcal/mol and -5.58Kcal/mol, respectively. It is important to note that while fluconazole was used as a control for the in silico analysis (Fig. 4), in the in vitro studies, paromomycin was chosen as the control compound, as it is the standard drug used for killing Leishmania parasites. This distinction reflects the different objectives of the in silico and in vitro analyses. This common mechanism of action makes them promising candidates for MD simulation and in vitro anti-parasitic assessments.
| Ligand | Binding energy (Kcal/mol) | Number of hydrogen bonds | Binding amino acid residues throgh hydrogen bonds |
|---|---|---|---|
| Posaconazole | -10.8 | 5 | TYR 115, HIS 420, LYS 421, CYS 422, TYR 456 |
| Isavuconazole | -9.3 | 3 | TYR 115 (two H-bonds), LYS 421 |
| Fluconazole (control) | -7.1 | 4 | HIS 420 (two H-bonds), ARG 360, TYR 115 |
TYR, tyrosine; HIS, histidine; ARG, arginine; CYS, cysteine; LYS, lysine
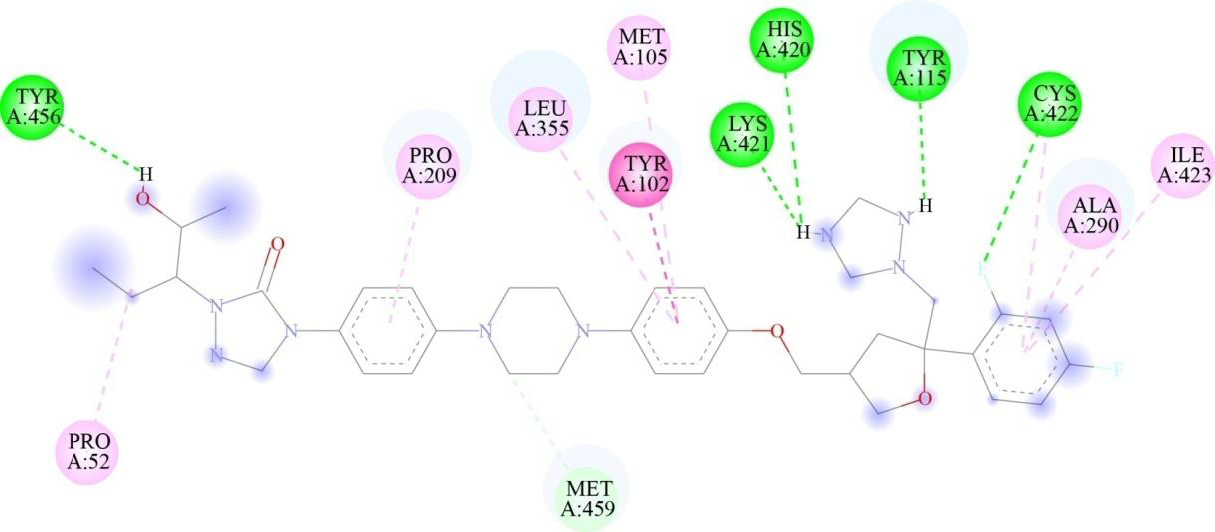
- Two-dimensional representation of binding interactions of posaconazole with the CYP51 protein of L. major: conventional H-bonds (green), carbon H-bonds (light green), alkyl (pink), Pi-alkyl (purple), and Pi Pi stacking (magenta) interactions.
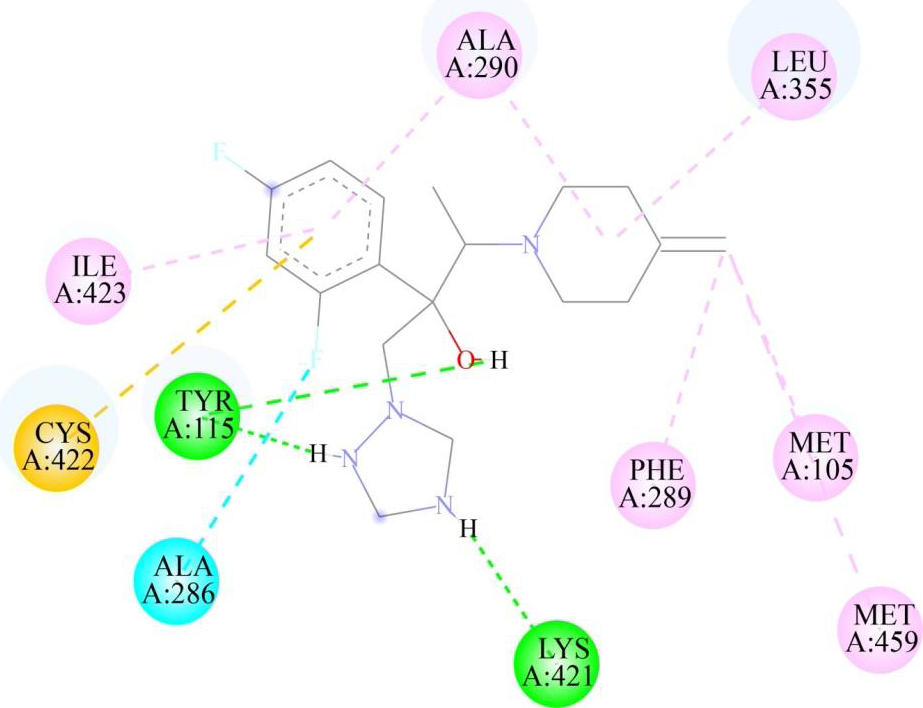
- Two-dimensional representation of binding interactions of isavuconazole with the CYP51 protein of L. major: green (H-bond interaction), light green (Carbon hydrogen interaction, cyan (Halogen interaction), orange (Pi-Sulfur interaction), purple (Alkyl interaction), pink (Pi-Alkyl interaction).
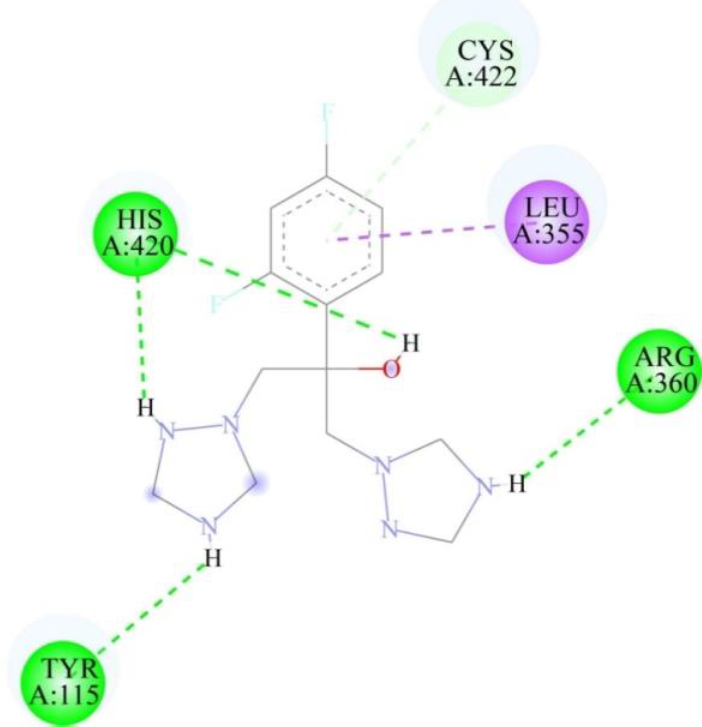
- Two-dimensional representation of binding interactions of fluconazole with the CYP51 protein of L. major: green (conventional H-bond interaction), light green (Pi-Donor H-bond interaction), and purple (Pi Sigma interaction).
Trajectories of molecular dynamic (MD) simulations
The 100 ns simulation revealed stable conformations for the CYP51 bound to posaconazole and isavuconazole, with Cα-backbone RMSD values ranging from 1.6 to 3.8 A (Fig. 5). The posaconazole- CYP51 complex remained stable, exhibiting minor fluctuations and a low RMSD value. However, between 70 ns and 100 ns, the complex underwent moderate conformational changes, becoming more flexible. The trajectory plot of the isavuconazole- CYP51 complex indicated an initial movement together for the first 10 ns, followed by separation, with the ligand's plot running below the protein's plot. This suggested that the ligand may experience significant conformational changes or explore a different binding site region than the protein. In contrast, the CYP51 protein showed a stable conformation throughout most of the simulation period when complexed with the control drug fluconazole. RMSF study revealed protein dynamics during drug binding. In MD simulations of posaconazole- CYP51 and isavuconazole- CYP51 complexes, the protein mostly maintained stable and rigid conformations (RMSF values <3), with select residues showing higher RMSF values up to 4.5, suggesting increased flexibility in regions like loops or binding sites undergoing conformational changes. Similarly, the control complex, fluconazole-CYP51, showed an overall stable conformation, with some regions displaying flexibility, likely corresponding to loops or binding regions interacting with the drug (Fig. 6).
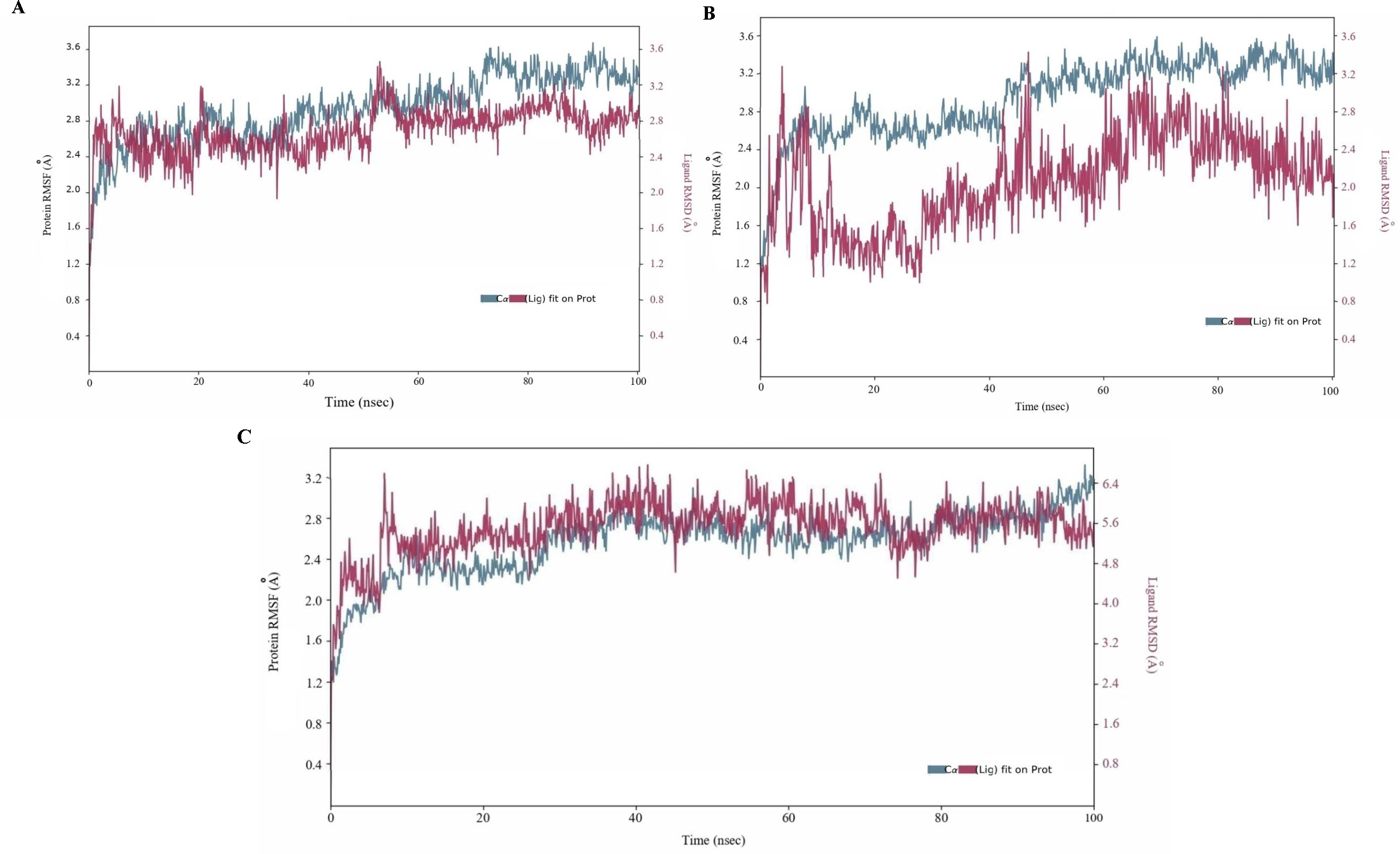
- MD simulation analysis of 100 ns trajectories of Cα-backbone backbone RMSD of CYP51 with (A) posaconazole, (B) isavuconazole and (C) fluconazole.
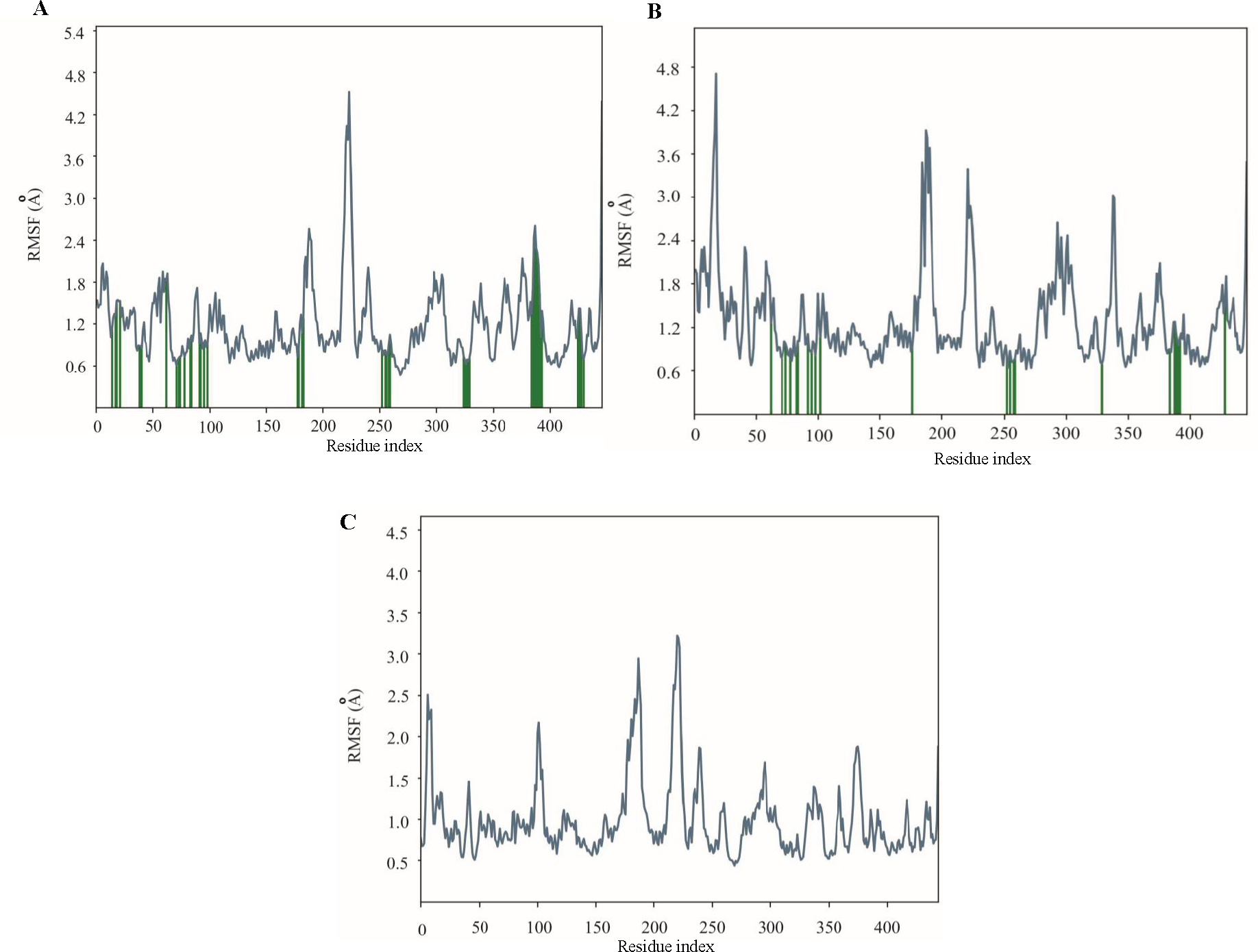
- MD simulation analysis of 100 ns trajectories of RMSF for CYP51 when complexed with (A) posaconazole, (B) isavuconazole, and (C) fluconazole.
In posaconazole-CYP51 simulations, significant interactions occurred, with CYS 422 and ARG 360 forming hydrogen bonds 43 per cent and 18 per cent of the time, respectively. Hydrophobic interactions engaged multiple residues for over 50 per cent of the simulation, and occasional water bridges were observed. Similarly, in isavuconazole-CYP51 simulations, stable interactions were noted, with ARG 114 and HIS 420 forming hydrogen bonds with the ligand for approximately 25 per cent and 40 per cent of the simulation, respectively. Hydrophobic interactions involved 11 residues, while no ionic interactions were evident. In contrast, fluconazole-CYP51 simulations revealed stronger hydrogen bonding interactions, with HIS 420, CYS 422, ILE 423, and TYR 115 forming H-bonds with fluconazole for approximately 95, 18, 10, and 4 per cent of the simulation period, respectively. Additionally, 17 amino acid residues were involved in hydrophobic interactions with the control drug. Water bridges were observed for a small fraction of the simulation. Compared to the control, both posaconazole and isavuconazole formed fewer hydrogen bonds. Still, they exhibited a wider range of hydrophobic interactions, suggesting potential differences in the stability and flexibility of the complexes (Fig. 7).
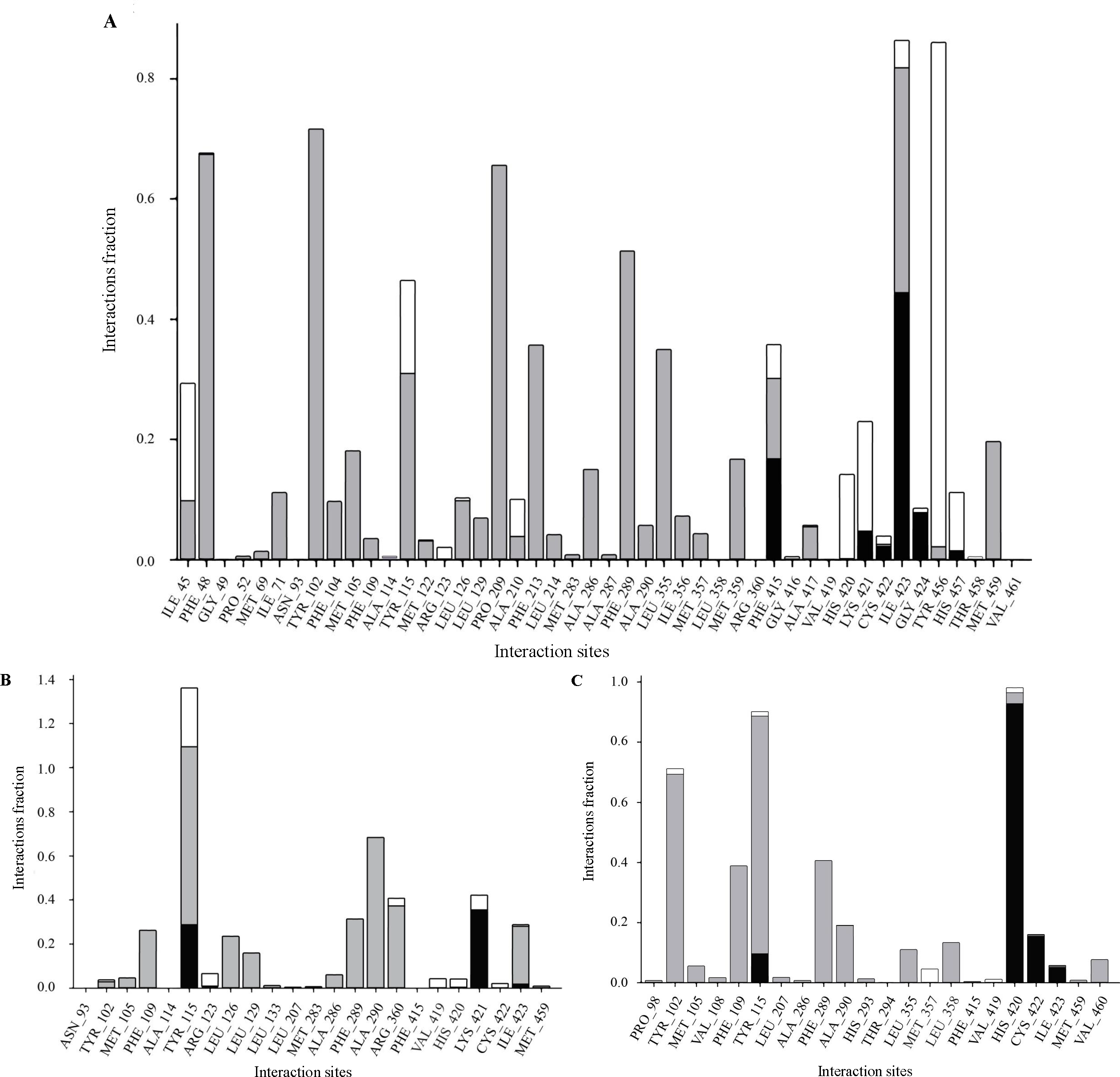
- Protein-Ligand contact for CYP51 with (A) posaconazole, (B) isavuconazol, and (C) fluconazole with grey bars representing hydrophobic interactions, black for hydrogen bonds, and white for water bridges.
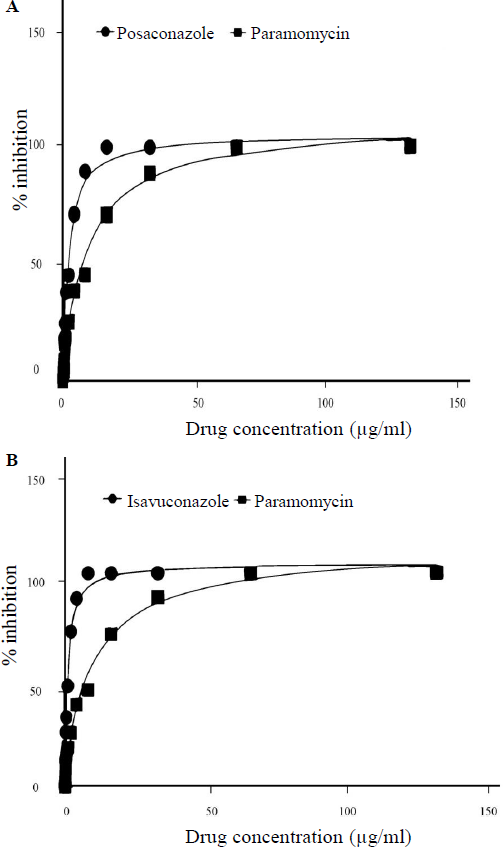
- Dose-response curves showing per cent inhibition of Leishmania growth: (A) Posaconazole vs. Paromomycin, (B) Isavuconazole vs. Paromomycin.
Evaluation of antileishmanial activity of posaconazole and isavuconazole in L. major promastigotes through IC50
The antileishmanial effect of posaconazole and isavuconazole against exponentially grown L. major (strain MHOM/SU/73/5ASKH) promastigotes was examined by an MTT assay. Treatment with posaconazole and isavuconazole (0-150 μg/ml) showed a reduction in parasite growth after 72h of incubation. The IC50 values for posaconazole and isavuconazole were calculated with 95 per cent confidence interval, yielding 2.062±0.89 µg/ml and 1.202±0.65 µg/ml, respectively indicating their potency in inhibiting the growth of promastigotes. The positive control drug, paromomycin, exhibited an IC50 value of 9.420±2.04 µg/ml. These findings demonstrate that both test drugs were effective in inhibiting the growth of L. major promastigotes, with isavuconazole showing slightly greater potency.
Discussion
Cutaneous Leishmaniasis (CL) poses a significant public health threat in regions like South America, the Middle East, Central Asia, and parts of Africa, where poverty and limited healthcare exacerbate its spread via sandfly bites42. Currently, treating cutaneous leishmaniasis has been challenging, with limited options available. Chemotherapy using drugs such as antimonials, paromomycin, miltefosine, and liposomal amphotericin B has been the mainstay, despite their drawbacks including toxicity profiles, potentially higher cost and the emergence of drug resistance43. Given these limitations, there is a critical need for innovative therapeutic strategies to address cutaneous leishmaniasis effectively. Hence, in this study, we tried to repurpose FDA-approved drugs against the CYP51 enzyme of L. major with the help of virtual screening and molecular docking complemented by MD simulations to validate the findings. Additionally, an in vitro study was conducted to assess the efficacy of the selected drugs against the parasite survival.
The enzyme CYP51 was targeted, which plays a pivotal role in L. major growth by facilitating the formation of membrane components. While the critical role of CYP51 for parasite survival in L. major remains debatable, research by McCall et al44 in 2015, employing gene knockout and pharmacological inhibition experiments, has demonstrated its indispensable role in the closely related species L. donovani. Another study by Xu et al45 in 2014 proposed that while CYP51 might not be vital for L. major's survival, its deficiency incurs a substantial fitness penalty, suggesting its essentiality and pivotal role in the parasite's biology and pathogenicity. The lesser similarity of this enzyme with the human proteome also makes it more suitable for use as a drug target, as off-target effects can be avoided.
From a collection of 1615 FDA-approved drugs screened against the modeled CYP51 enzyme, posaconazole (-10.8 kcal/mol with 5 hydrogen bonds) and isavuconazole (-9.3 kcal/mol with 3 hydrogen bonds) exhibited high binding energies and favourable interactions. Recognized for their efficacy against the fungal enzyme CYP51 and sharing approximately 30 per cent similarity with it, posaconazole and isavuconazole were selected for further molecular dynamics (MD) simulations.
In 100 nano second simulations, both CYP51 complexes with posaconazole and isavuconazole initially exhibited stable conformations but demonstrated moderate conformational changes towards the end. The RMSF plots for both posaconazole and isavuconazole show elevated RMSF values in specific regions, suggesting that these areas may correspond to the proteins' loop regions or terminal ends, which are typically more flexible. The MD simulations confirmed the protein-ligand contacts identified in the docking studies, validating the binding residues for the respective drugs as predicted. These contacts lie within the predicted binding sites, further supporting their relevance. Furthermore, the MD simulations revealed additional amino acid residues interacting with the ligands compared to the molecular docking results, indicating a broader binding mode and underscoring the dynamic nature of the binding site through additional contacts (Table IV). In contrast, the MD simulation of the control drug fluconazole with CYP51 showed a stable conformation, with minimal changes throughout the simulation. Hydrogen bonds with HIS 420 and CYS 422 were maintained for 95 per cent and 18 per cent of the simulation, indicating a more robust and consistent binding pattern. Fluconazole also formed more hydrophobic interactions (with 17 residues), suggesting a stable binding mode.
| Parameters | Residues | ||
|---|---|---|---|
| Binding site predicted | LEU_126, LEU_133, ALA_287, GLY_291, THR_294, ILE_349, PRO_354, LEU_355, LEU_358, ARG_360, PHE_415, HIS_420, CYS_422, ILE_423 | ||
| Parameters | Posaconazole | Isavuconazole | Fluconazole |
| Molecular docking | PRO_52, TYR_102, MET_105, TYR_115, PRO_209, ALA_290, LEU_355, HIS_420, LYS_421, CYS_422, ILE_423, TYR_456, MET_459 | MET_105, TYR_115, ALA_286, PHE_289, ALA_290, LEU_355, LYS_421, CYS_422, ILE_423, MET_459 | TYR_115, LEU_355, ARG_360, HIS_420, CYS_422 |
| Molecular dynamic simulations | ILE_45, PHE_48, GLY_49, PRO_52, MET_69, ILE_71, ASN_93, TYR_102, PHE_104, MET_105, PHE_109, ALA_114, TYR_115, MET_122, ARG_123, LEU_126, LEU_129, PRO_209, ALA_210, PHE_213, LEU_214, MET_283, ALA_286, ALA_287, PHE_289, ALA_290, LEU_355, ILE_356, MET_357, LEU_358, MET_359, ARG_360, PHE_415, GLY_416, ALA_417, VAL_419, HIS_420, LYS_421, CYS_422, ILE_423, GLY_424, TYR_456, HIS_457, THR_458, MET_459, VAL_461, | ASN_93, TYR_102, MET_105, PHE_109, ALA_114, TYR_115, ARG_123, LEU_126, LEU_129, LEU_133, LEU_207, MET_283, ALA_286, PHE_289, ALA_290, ARG_360, PHE_415, VAL_419, HIS_420, LYS_421, CYS_422, ILE_423, MET_459, | PRO_98, TYR_102, MET_105, VAL_108, PHE_109, TYR_115, LEU_207, ALA_286, PHE_289, ALA_290, HIS_293, THR_294, LEU_355, MET_357, LEU_358, PHE_415, VAL_419, HIS_420, CYS_422, ILE_423, MET_459, VAL_460 |
LEU, leucine; ALA, alanine; GLY, glycine; THR, threonine; ILE, isoleucine; PRO, proline; PHE, phenylalanine; MET, methione; VAL, valine; ASN, asparagines; HIS, histidine; CYS, cysteine; ARG, arginine
Posaconazole is a triazole antifungal with a complex structure, characterized by multiple chiral centers that enhance its potent inhibition of 14-alpha demethylase, making it highly effective in treating severe systemic infections. Its stereochemistry facilitates strong binding interactions, essential for its broad-spectrum activity46,47. In contrast, isavuconazole features a butan-2-ol backbone with various functional groups, including 1,2,4-triazol-1-yl, 2,5-difluorophenyl, and 4-(p-cyanophenyl)-1,3-thiazol-2-yl groups. These structural components enable it to disrupt cell membranes by reducing ergosterol levels48,49. Several studies have explored the use of different azole drugs to treat leishmaniasis. A study conducted by Shakya et al50 in 2011, showcasing the combined use of ketoconazole and miltefosine with the immunomodulator picroliv against visceral leishmaniasis, suggested the potency of ketoconazole in combination therapy in treating leishmaniasis. Similarly, other studies have employed the use of fluconazole and itraconazole to target cutaneous leishmaniasis caused by L. major51,52. Our study suggested posaconazole and isavuconazole against the CYP51 enzyme of L. major. Posaconazole and isavuconazole, being broad-spectrum triazoles with enhanced bioavailability, possess a notable advantage53,54. In vitro study demonstrated that posaconazole and isavuconazole effectively inhibited the growth of L. major promastigotes, with respective IC50 values of 2.062±0.89 µg/ml and 1.202±0.65 µg/ml, respectively, suggesting their efficacy as anti-leishmanial agents.
In the present study, we observed that both posaconazole and isavuconazole act against the CYP51 enzyme of L. major. This was demonstrated by molecular docking and MD simulation studies. Additionally, an in vitro study using L. major strains suggested that both drugs can effectively kill the parasite's promastigote form. Therefore, these two drugs can potentially be repurposed for the treatment of cutaneous leishmaniasis.
However, there are several limitations to our study. Firstly, while the in vitro results are promising, the experiments were conducted only on the promastigote form of L. major, and the effects on the clinically relevant amastigote form remain to be determined. Additionally, although molecular docking studies suggest that these drugs target the CYP51 enzyme, further validation is required to confirm specific enzyme interactions in the parasite. To address these limitations, future research should focus on testing the efficacy of both drugs against the amastigote form of L. major in in vivo models. Additionally, conducting animal experiments would be essential to assess pharmacokinetics, toxicity, and drug efficacy in a living organism. Moreover, membrane-bound MD simulations in future studies could provide deeper insights into drug permeability and efficacy within a more physiologically relevant context. Finally, based on the individual efficacy of these drugs, future studies should investigate their synergistic potential to enhance therapeutic outcomes.
Acknowledgment
Authors acknowledge the department of Medical Parasitology, Post Graduate Institute of Medical Education and Research Chandigarh, for providing the necessary facilities to carry out this research. Author also thanks to Dr. Harshita Sharma for her dedicated efforts in maintaining the Leishmania culture throughout the experiment. We would also like to thank to Mr. George Fernandez Fauche for supporting in graphics.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author upon request.
Financial support & sponsorship
None.
Conflicts of Interest
None.
Use of Artificial Intelligence (AI)-Assisted Technology for manuscript preparation
The authors confirm that there was no use of AI-assisted technology for assisting in the writing of the manuscript and no images were manipulated using AI.
References
- A Review of Leishmaniasis: Current knowledge and future directions. Curr Trop Med Rep. 2021;8:121-32.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Unusual observations in Leishmaniasis-An Overview. Pathogens. 2023;12:297.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- From infection to death: An overview of the pathogenesis of Visceral Leishmaniasis. Pathogens. 2023;12:969.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Visceral leishmaniasis: A global overview. J Glob Health Sci. 2020;2:e3.
- [CrossRef] [Google Scholar]
- Cutaneous and mucocutaneous leishmaniasis. Dermatol Ther. 2009;22:491-502.
- [CrossRef] [PubMed] [Google Scholar]
- Cutaneous and mucocutaneous leishmaniasis: Experience of a Mediterranean hospital. Parasit Vectors. 2020;13:24.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Leishmaniasis. Available from: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis, accessed on February 21, 2024.
- Role of efflux pumps and intracellular thiols in natural antimony resistant isolates of Leishmania donovani. PLoS One. 2013;8:e74862.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The role of ATP-binding cassette transporter genes expression in treatment failure cutaneous leishmaniasis. AMB Express. 2022;12:78.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Evaluation of MDR1 and MRPA genes expression in different types of dry cutaneous leishmaniasis. BMC Res Notes. 2019;12:803.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Investigation of the pathways related to intrinsic miltefosine tolerance in Leishmania (Viannia) braziliensis clinical isolates reveals differences in drug uptake. Int J Parasitol Drugs Drug Resist. 2019;11:139-47.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Genomic Appraisal of the multifactorial basis for in vitro acquisition of miltefosine resistance in Leishmania donovani. Antimicrob Agents Chemother. 2016;60:4089-100.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Genomic instability at the locus of sterol C24-methyltransferase promotes amphotericin B resistance in Leishmania parasites. PLoS Negl Trop Dis. 2019;13:e0007052.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Sterol 14α-demethylase mutation leads to amphotericin B resistance in Leishmania mexicana. PLoS Negl Trop Dis. 2017;11:e0005649.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Amphotericin B resistance in Leishmania mexicana: Alterations to sterol metabolism and oxidative stress response. PLoS Negl Trop Dis. 2022;16:e0010779.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- An insight into the current perspective and potential drug targets for Visceral Leishmaniasis (VL) Curr Drug Targets. 2020;21:1105-29.
- [CrossRef] [PubMed] [Google Scholar]
- Exploring Leishmania donovani 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) as a potential drug target by biochemical, biophysical and inhibition studies. Microb Pathog. 2014;66:14-23.
- [CrossRef] [PubMed] [Google Scholar]
- Metabolic pathways of Leishmania Parasite: Source of pertinent drug targets and potent drug candidates. Pharmaceutics. 2022;14:1590.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Identification of selective inhibitors of Ld DHFR Enzyme using pharmacoinformatic methods. J Comput Biol. 2021;28:43-59.
- [CrossRef] [PubMed] [Google Scholar]
- In silico identification and in vitro evaluation of natural inhibitors of leishmania major Pteridine Reductase I. Molecules. 2017;22:2166.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Ammonium trichloro [1,2-ethanediolato-O,O']-tellurate cures experimental visceral leishmaniasis by redox modulation of Leishmania donovani trypanothione reductase and inhibiting host integrin linked PI3K/Akt pathway. Cell Mol Life Sci. 2018;75:563-88.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Molecular events leading to death of Leishmania donovani under spermidine starvation after hypericin treatment. Chem Biol Drug Des. 2017;90:962-71.
- [CrossRef] [PubMed] [Google Scholar]
- L-Asparaginase as a new molecular target against leishmaniasis: Insights into the mechanism of action and structure-based inhibitor design. Mol Biosyst. 2015;11:1887-96.
- [CrossRef] [PubMed] [Google Scholar]
- Targeting ergosterol biosynthesis in Leishmania donovani: Essentiality of sterol 14 alpha-demethylase. PLoS Negl Trop Dis. 2015;9:e0003588.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Sterol 14α-demethylase from trypanosomatidae parasites as a promising target for designing new antiparasitic agents. Curr Top Med Chem. 2021;21:1900-21.
- [CrossRef] [PubMed] [Google Scholar]
- Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022;50:D20-D26.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The protein data bank. Nucleic Acids Res. 2000;28:235-42.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480-9.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296-W303.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477-86.
- [CrossRef] [PubMed] [Google Scholar]
- Accelrys Inc. Discovery Studio Visualizer (Version 21.1.0). Dassault Systèmes, 2022. Available from: https://discover.3ds.com/discovery-studio-visualizer, accessed on November 29, 2022.
- PrankWeb: A web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019;47:W345-9.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- ProBiS-ligands: A web server for prediction of ligands by examination of protein binding sites. Nucleic Acids Res. 2014;42:W215-20.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- COACH-D: Improved protein-ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res. 2018;46:W438-42.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- ZINC: A free tool to discover chemistry for biology. J Chem Inf Model. 2012;52:1757-68.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Open Babel: An open chemical toolbox. J Cheminform. 2011;3:33.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243-50.
- [CrossRef] [PubMed] [Google Scholar]
- AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785-91.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Computational insights into tetracyclines as inhibitors against SARS-CoV-2 Mpro via combinatorial molecular simulation calculations. Life Sci. 2020;257:118080.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Discovery of Novel hsp27 inhibitors as prospective anti-cancer agents utilizing computer-assisted therapeutic discovery approaches. Cells. 2022;11:2412.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Evidence for reversal of immunosuppression by homeopathic medicine to a predominant Th1-type immune response in BALB/c mice infected with Leishmania donovani. Homeopathy. 2022;111:31-41.
- [CrossRef] [PubMed] [Google Scholar]
- Epidemiology of Leishmaniasis. In: Almeida-Souza F, Cardoso F de O, Abreu-Silva AL, Calabrese K da S, eds. Leishmania parasites-epidemiology, immunopathology and hosts. London, UK: IntechOpen; 2024.
- [Google Scholar]
- An appraisal of the scientific current situation and new perspectives in the treatment of cutaneous leishmaniasis. Acta Trop. 2021;221:105988.
- [CrossRef] [PubMed] [Google Scholar]
- Targeting Ergosterol biosynthesis in Leishmania donovani: Essentiality of sterol 14 alpha-demethylase. PLoS Negl Trop Dis. 2015;9:e0003588.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Sterol biosynthesis is required for heat resistance but not extracellular survival in leishmania. PLoS Pathog. 2014;10:e1004427.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Clinical pharmacodynamics and pharmacokinetics of the antifungal extended-spectrum triazole posaconazole: An overview. Br J Clin Pharmacol. 2010;70:471-80.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Pharmacokinetics and Pharmacodynamics of Posaconazole. Drugs. 2020;80:671-95.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- New perspectives on antimicrobial agents: Isavuconazole. Antimicrob Agents Chemother. 2022;66:e0017722.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- PubChem 2023 update. Nucleic Acids Res. 2023;51:D1373-80.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Successful treatment of Old World cutaneous leishmaniasis caused by Leishmania infantum with posaconazole. Antimicrob Agents Chemother. 2011;55:1774-6.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Fluconazole for the treatment of cutaneous leishmaniasis caused by Leishmania major. N Engl J Med. 2002;346:891-5.
- [CrossRef] [PubMed] [Google Scholar]
- A randomized, double-blind, placebo-controlled clinical trial of itraconazole in the treatment of cutaneous leishmaniasis. J Eur Acad Dermatol Venereol. 2005;19:80-3.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical research advances of isavuconazole in the treatment of invasive fungal diseases. Front Cell Infect Microbiol. 2022;12:1049959.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Review of the alternative therapies for onychomycosis and superficial fungal infections: Posaconazole, fosravuconazole, voriconazole, oteseconazole. Int J Dermatol. 2022;61:1431-41.
- [CrossRef] [PubMed] [Google Scholar]




