
Translate this page into:
Novel mutations of the arylsulphatase B (ARSB) gene in Indian patients with mucopolysaccharidosis type VI
Reprint requests: Dr Ashwin B. Dalal, Diagnostics Division, Centre for DNA Fingerprinting & Diagnostics 4-1-714, Tuljaguda Complex, Mozamzahi Road, Nampally, Hyderabad 500 001, Telangana, India e-mail: ashwindalal@gmail.com
-
Received: ,
This is an open access article distributed under the terms of the Creative Commons Attribution NonCommercial ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
Mucopolysaccharidosis type VI (MPS VI) is a rare, autosomal recessive lysosomal storage disorder caused by deficient enzymatic activity of N-acetyl galactosamine-4-sulphatase resulting from mutations in the arylsulphatase B (ARSB) gene. The ARSB gene is located on chromosome 5q11-q13 and is composed of eight exons. More than hundred ARSB mutations have been reported so far, but the mutation spectrum of MPS VI in India is still unknown. Hence, the aim of the present study was to identify the mutational spectrum in patients with MPS VI in India and to study the genotype-phenotype association and functional outcomes of these mutations.
Methods:
Molecular characterization of the ARSB gene by Sanger sequencing was done for 15 patients (aged 15 months to 11 yr) who were enzymatically confirmed to have MPS VI. Age of onset, clinical progression and enzyme activity levels in each patient were studied to look for genotype-phenotype association. Haplotype analysis performed for unrelated patients with the recurring mutation W450C, was suggestive of a founder effect. Sequence and structural analyses of the ARSB protein using standard software were carried out to determine the impact of detected mutations on the function of the ARSB protein.
Results:
A total of 12 mutations were identified, of which nine were novel mutations namely, p.D53N, p.L98R, p.Y103SfsX9, p.W353X, p.H393R, p.F166fsX18, p.I220fsX5, p.W450L, and p.W450C, and three were known mutations (p.D54N, p.A237D and p.S320R). The nine novel sequence variants were confirmed not to be polymorphic variants by performing sequencing in 50 unaffected individuals from the same ethnic population.
Interpretation & conclusions:
Nine novel mutations were identified in MPS VI cases from India in the present study. The study also provides some insights into the genotype-phenotype association in MPS VI.
Keywords
ARSB gene
ARSB protein structural analysis
genotype-phenotype association
Indian patients
mucopolysaccharidosis type VI
mutations

Mucopolysaccharidoses (MPS) are a group of rare inherited metabolic disorders caused by absence or inadequate functioning of lysosomal enzymes required to break down complex molecules called glycosaminoglycans (GAGs). This group includes around seven disorders, each caused by deficiency of one of the enzymes involved in the catabolism of mucopolysaccharides (MPS type I, II, III, IV, VI, VII and IX)1. MPS VI (Maroteaux-Lamy syndrome) is caused by deficient enzymatic activity of N-acetyl galactosamine-4-sulphatase otherwise known as arylsulphatase B, which belongs to the sulphatase family of enzymes. This deficiency is caused by mutations in the ARSB gene which encodes this enzyme. MPS VI is an autosomal recessive lysosomal storage disorder, characterized by the accumulation of dermatan sulphate in tissues and its increased excretion in urine12. The incidence of MPS VI varies among different populations and the birth prevalence ranges from 1 in 50,000 to 1 in 1.5 million live births3. Maroteaux-Lamy syndrome has a spectrum of manifestations which include coarse facial features, clouded cornea, hepatosplenomegaly, dysostosis multiplex and stunted growth. In most cases of MPS VI the intelligence is normal and prominent metachromatic inclusions may be seen in leucocytes4. Enzyme replacement therapy (ERT) with recombinant human arylsulphatase B (galsulphase) is available for MPS VI and has been documented to improve the cardiac and pulmonary functions in affected individuals3. The ARSB gene is located on chromosome 5q11-q13 and is composed of eight exons. More than hundred ARSB mutations have been reported so far but its mutation spectrum in India is still unknown4. We undertook this study to identify the mutations in Indian patients with MPS VI and to further evaluate the genotype-phenotype association of these mutations.
Material & Methods
Sample selection: A total of 15 patients with MPS VI were included in the study. The inclusion of each patient was done following complete clinical evaluation by a qualified clinical geneticist, urinary glycosaminoglycan assay through cellulose acetate gel electrophoresis and confirmation of deficient enzyme activity by spectrophotometric assay of arylsuphatase B enzyme activity in peripheral blood leucocytes. While the patients were referred from different clinical centres of India during January 2011 to July 2013, the molecular genetic study was conducted in the Diagnostics Division of the Centre for DNA Fingerprinting and Diagnostics (CDFD), Hyderabad, India. As all the patients were under 18 yr of age (15 months to 11 yr), written informed consent was obtained from either a parent or a legal guardian. Ethical clearance of the research protocol was obtained from the Institute Ethics Committee of CDFD. Ten millilitres of urine (for urine glycosaminoglycan assay), 5 ml of peripheral blood anticoagulated with potassium-EDTA (for ARSB enzyme assay and for DNA isolation followed by mutation analysis) and 5 ml of peripheral blood anticoagulated with lithium heparin (for establishing immortal lymphoblastoid cell lines) were collected from each patient. In addition, 5 ml of peripheral blood anticoagulated with potassium-EDTA was collected from the parents and affected relatives and unaffected siblings (for enzyme assay and mutation analysis), wherever possible.
Lysosomal ARSB enzyme assay: Arylsulphatase B activity was measured spectrophotometrically in the peripheral blood leucocytes using the artificial substrate p-nitrocatechol sulphate5. Protein concentration was also determined6. The homogenates were incubated in the presence of the substrate for one hour and the residual enzymatic activity was expressed as percentage of normal values.
Mutation analysis of ARSB gene: Genomic DNA was extracted using salting out method7 and the phenol-chloroform technique8 from the peripheral blood samples of all 15 patients diagnosed with MPS VI as well as from the samples of their family members. Primers were designed for all the eight exons and flanking exon-intron boundaries of ARSB using the Primer3 Input version 0.4.09 (http://bioinfo.ut.ee/primer3-0.4.0/) and NCBI primer BLAST softwares (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) (National Library of Medicine, Bethesda, USA). Polymerase chain reaction (PCR) was carried out in the BIORAD DNAEngine Peltier Thermal Cycler (Applied Biosystems, USA) with the designed primers for the ARSB gene (Table I). PCR was carried out for 30 cycles with an initial denaturation for 8 min at 96°C and final extension for 7 min at 72° C (Table I). The total reaction mixture (25 µl) contained 100ng DNA template, 1X PCR buffer (Fermentas Life sciences, Lithuania), 2.0 nmol/l MgCl2, 0.2µmol/l of each dNTP, 10 pmol of each primer, and 1 U of Taq polymerase (5 units/µl GeneTAQ Recombinant Taq DNA Polymerase, (Nippon Gene Co. Ltd., Tokyo). PCR products were purified either using Qiagen PCR Purification kit (QIAquick, Qiagen, Hilden, Germany) or by gel elution using the QIAquick gel extraction kit (Qiagen, Hilden, Germany). Bidirectional Sanger sequencing was carried out on all the purified PCR products on the ABI 3130 automated genetic analyser (Applied Biosystems, USA). Sequencing data were analysed using the sequence analysis software Chromas Lite (Technelysium pty Ltd, Australia), and pair-wise alignment of both the reference exonic sequence and the patient's PCR amplified exonic sequences was carried out using the EMBOSS (http://www.ebi.ac.uk/Tools/psa/emboss_needle/nucleotide.html) (The European Bioinformatics Institute [EMBL-EBI], United Kingdom) nucleotide alignment tool for the identification of the sequence variants.
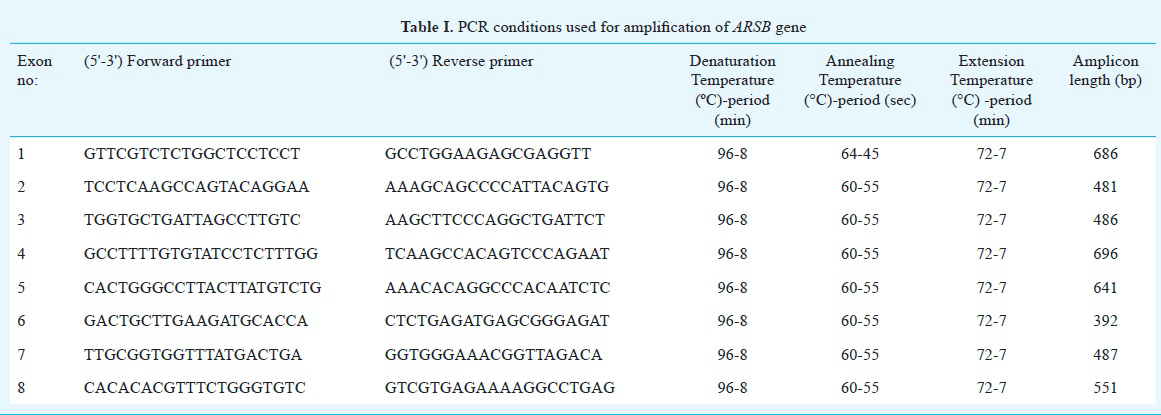
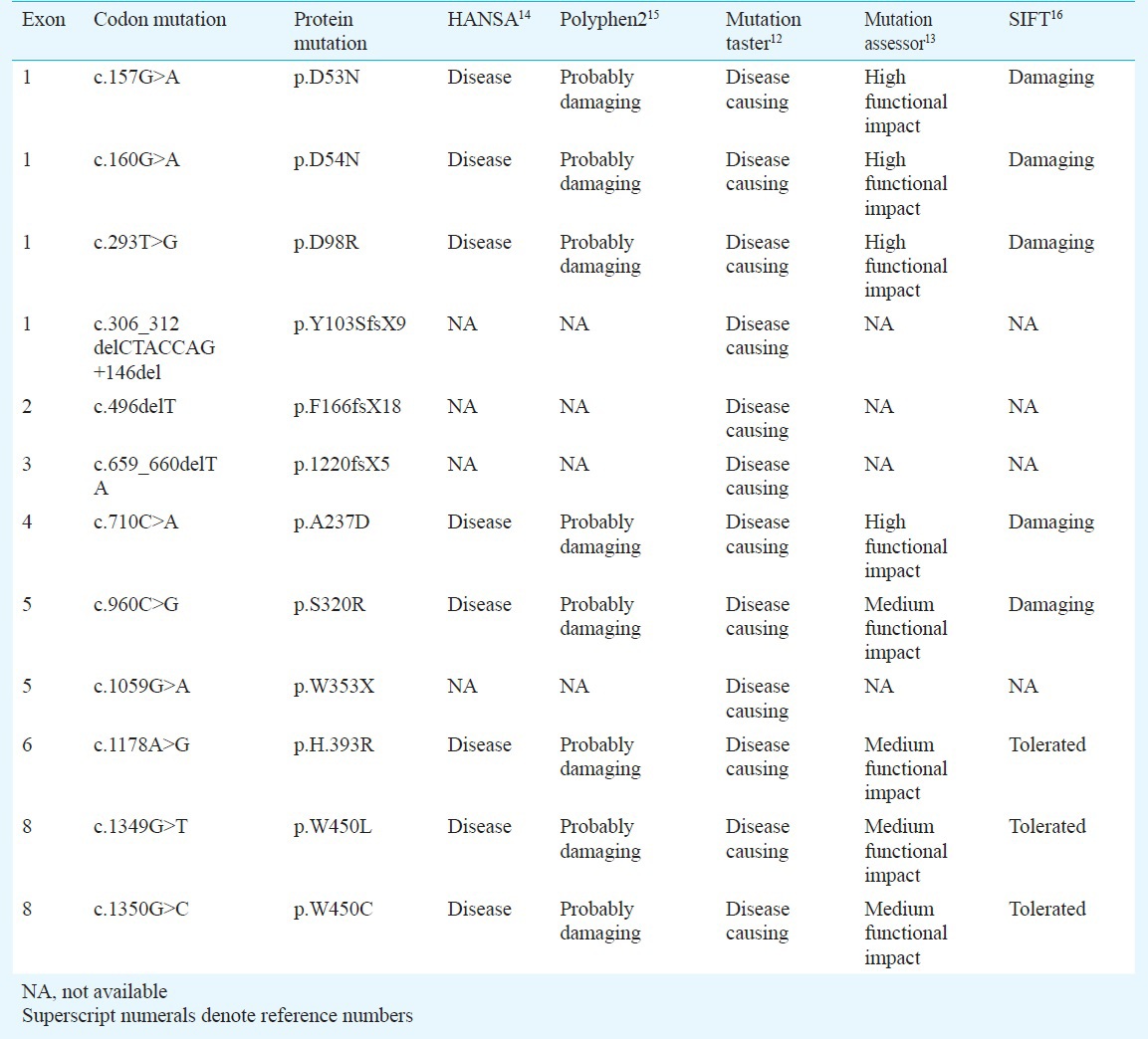
Characterization of novel mutations in the ARSB gene: The sequence variations/mutations detected in the study were described using Human Genome Variation Society nomenclature1011 (HGVS - http://www.hgvs.org/mutnomen/). The detected novel sequence variants were considered to be mutations based on the following criteria: (i) absence of these novel mutations in 50 healthy individuals from the same ethnic group; (ii) exhibition of Mendelian segregation pattern in the family being studied; (iii) prediction of the disease causing potential of a missense mutation by the use of mutation prediction softwares like Mutation Taster12, (http://www.mutationtaster.org/), Mutation Assessor13, (http://mutationassessor.org/), HANSA14, Polyphen15, and SIFT16; (iv) evolutionary conservation of amino acid residues or motifs across different species17, (v) database search of the Human Genome Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/ac/index.php) for distinguishing known mutations from novel ones; and (vi) comparison with databases like dbSNP (http:www.ncbi.nlm.nih.gov/SNP/) for filtering out all the natural variants.
Haplotype analysis: For the two patients identified to have the homozygous mutation p.W450C, haplotype analysis was done using three different microsatellite markers18. The markers AFM2142G9, RH41120 and SGHC-104311 were located proximal, distal and intragenic to the ARSB gene. Haplotype analysis was also carried out for the three different unrelated patients in the study with the homozygous mutation p.L98R, using the same three markers. PCR with the fluorescently end-labelled primers (microsatellite markers) (BIORAD DNAEngine Peltier Thermal Cycler, Applied Biosystems, USA) was performed and fluorescent PCR products were genotyped by capillary electrophoresis on ABI 3130xl Automated Genetic Analyzer (Applied Biosystems, USA). The haplotype results obtained were analysed using the software GeneMarker V1.85 version (Softgenetics, USA).
Sequence and structural analysis: Homologous sequences of P15848 (protein product of ARSB gene) were obtained by running three rounds of PSIBLAST (National Library of Medicine, USA) (http://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE=Proteins&PROGRAM=blastp&RUN_PSIBLAST=on) against the non-redundant database. Multiple sequence alignments were generated using the homologous sequences having sequence coverage of at least 95 per cent of the query sequence with ClustalW-2.119 running on default parameters. Gribskov's score was calculated using the PROPHECY program of EMBOSS by incorporating the BLOSUM62 matrix20. The X-ray structure of the P15848 protein was also used for further analysis (PDB code 1FSU)21. The per cent solvent accessible surface areas of the residues were calculated using PSA program of the JOY suite (Fig. 1)22. TRANSEQ program of EMBOSS was used to obtain translated products of the deletion mutants c.1208delC and c.496delT23. PyMOL24 and Swiss-PdbViewer 4.0.425 were used for performing structural analysis and rendering figures.

-
ARSB sequence and structural analysis: Protein Data Bank (PDB) file output of JOY for protein P15848. Residues form alpha helices, beta strands and 310 helices are coloured red, blue and maroon, respectively. Solvent accessible and inaccessible residues are written in lower and upper cases, respectively. Residues that bind to the main-chain amide using hydrogen bonds are written in bold and the ones that bind to main-chain carbonyl are underlined. Residues with positive phi torsion angles are italicised and the ones that form disulphide bond have a cedilla. Positions of mutated residues have been enclosed in a purple box.
Results
A total of 15 patients with MPS VI including two siblings (from 14 different families) were studied. There were 12 male and three female patients and the age at presentation ranged from 15 months to 11 yr. These patients were diagnosed to have MPS VI based on their clinical features and arylsulfatase B enzyme activity levels (patients 1 to 15: Table II). Pedigree analysis showed the presence of consanguinity in ten families (11 patients) (Table II). There was considerable variation in the phenotypic manifestations. The phenotypes were classified into mild, intermediate and severe based on the age of onset, severity of clinical features, rate of progression of the disease and ARSB enzyme activity levels. Twelve of the 15 patients had a severe phenotype, one (patient no. 9) had an intermediate phenotype and two patients who were siblings (patient nos. 12 and 13) had a mild phenotype. None of the patients was given enzyme replacement therapy (ERT) because of financial constraints.
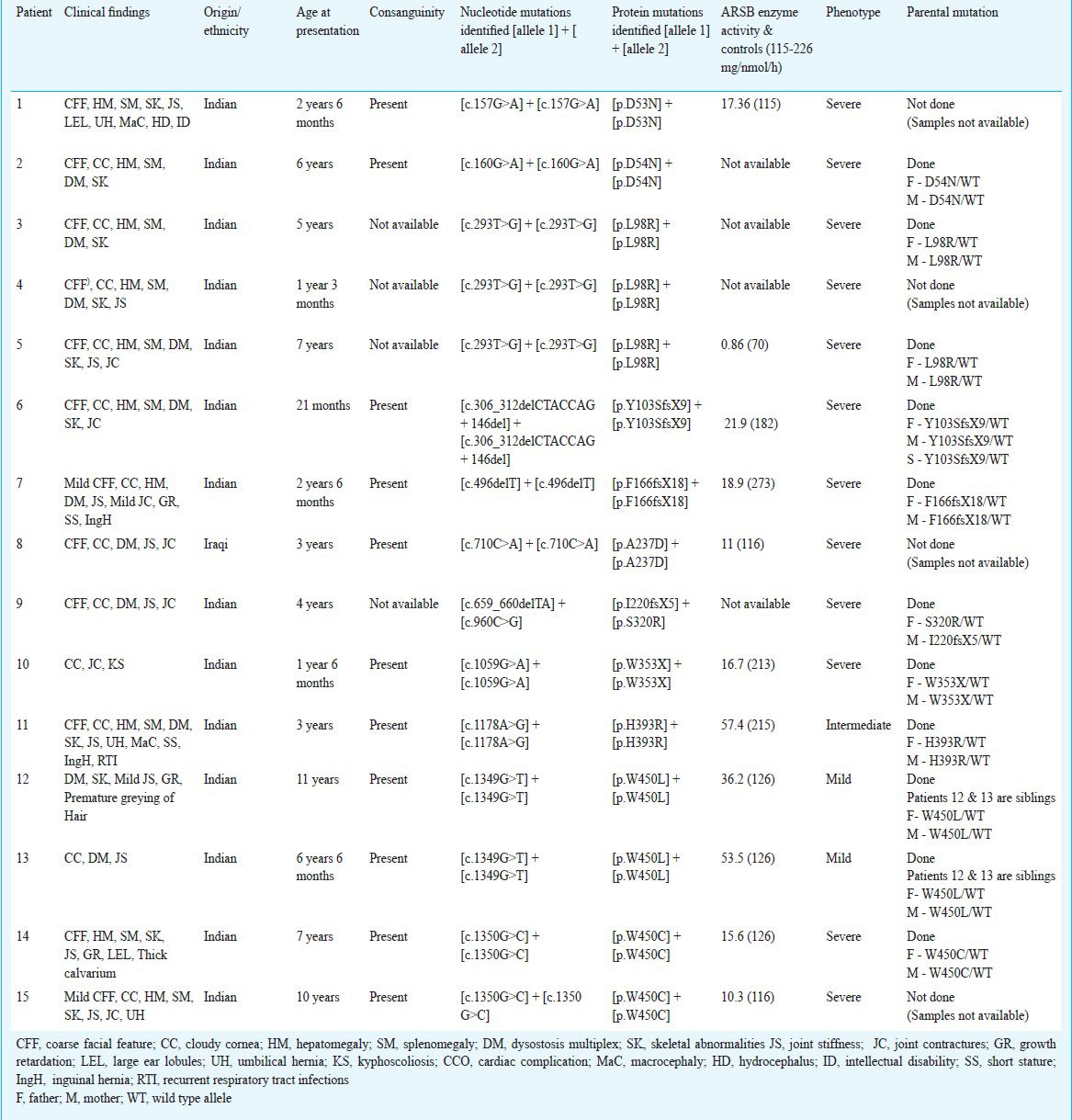
Molecular characterization of the ARSB gene was performed for identification of all the possible deleterious and potential disease causing mutations. The entire open reading frame of ARSB was PCR-amplified from genomic DNA isolated from the peripheral blood of MPS VI patients. A total of 12 different mutations (http://www.hgvs.org/mutnomen/recs.html)2122 were found in the 14 families (15 patients) Table II). Of the 12 detected mutations, nine viz. p.D53N, p.L98R, p.Y103SfsX9, p.F166fsX18, p.I220fsX5, p.W353X, p.H393R, p.S403Yfs, p.P445L, p.W450L, and p.W450C were novel, unreported mutations and three p.D54N (c.160G>A), p.A237D (c.710C>A), and p.S320R (c.960C>G) were known mutations (Table III). The novel sequence variations detected were analysed for their disease-causing potential with the help of pathogenicity-prediction softwares like HANSA, MutationTaster, Polyphen2, SIFT and Mutation Assessor (Table III). The results supported the assumption that these novel sequence variations were most likely to be disease-causing and deleterious rather than natural variants.
The missense mutations p.L98R, and p.W450C associated with a severe phenotype, were recurrently found in different unrelated MPS VI patients. The p.L98R mutation was present in three unrelated patients. The p.W450C mutation was present in two unrelated patients. Haplotypes of the two patients with p.W450C mutation in the present study were similar and showed homozygosity for all the three microsatellite markers (one intragenic and two flanking markers) analysed, suggesting that these patients might have originated from a common ancestor. However, haplotype analysis for the three patients with the L98R mutation with the same microsatellite markers, revealed different haplotypes, suggesting the absence of a founder effect and the likely possibility of it being a hotspot for mutation.
On extensive sequence and structural analysis (Fig. 1) of the identified mutations it was found that all the affected residues had a high degree of evolutionary conservation. Both position-specific profile Gribskov's score and multiple sequence alignments indicated that residues at the affected sites were highly conserved and any changes in these positions were expected to cause deleterious effects. In the protein all the residues at the identified affected sites, except tryptophan (W) at the 450th position, were buried.
The side chains of aspartate (Asp) residues at 53 and 54 positions were buried in the protein and formed coordination bonds with the Ca2+ ligand and were also part of the active site (Fig. 2). The residue L98 was in a helix and A237 was in a β-strand (Fig. 3a). The change of hydrophobic amino acid residue from Leu (a strong helix favouring residue) to positively charged Arg (a not so helix favouring residue) at position 98 could possibly affect the helical H-bonding thereby destabilizing the helix. The change of Ala to Asp position 237 could disrupt the β strand which forms part of a β-sheet. On modelling and analysing this mutation using Swiss-PdbViewer4.0.4, it was observed that Asp substitution at position 237 was stereochemically not feasible. Further, this position was in close proximity (within 6 Angstrom) of the active site residues Arg9 and Lys145 and, therefore, any alteration to Ala residue at position 237 could possibly affect the active-site conformation resulting in the disruption of enzymatic activity (Fig. 3a). Mutation from His to Arg at position 393 could also destabilize the β-sheet by disrupting intra β-strand h-bonding (Fig. 3b) with other residues.
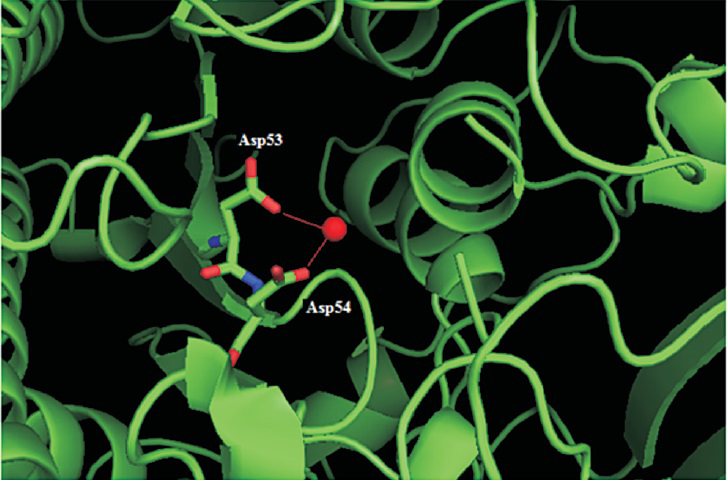
- Structural analysis of the mutations of ARSB gene: The Asp53 and Asp54 (residues are shown using the stick model) make coordination bonds with the ligand Ca2+ (CA604 shown as a red sphere). The Figure has been rendered using PyMOL (DeLano Scientific, San Carlos, CA) using the Protein Data Bank (PDB) entry 1FSU.
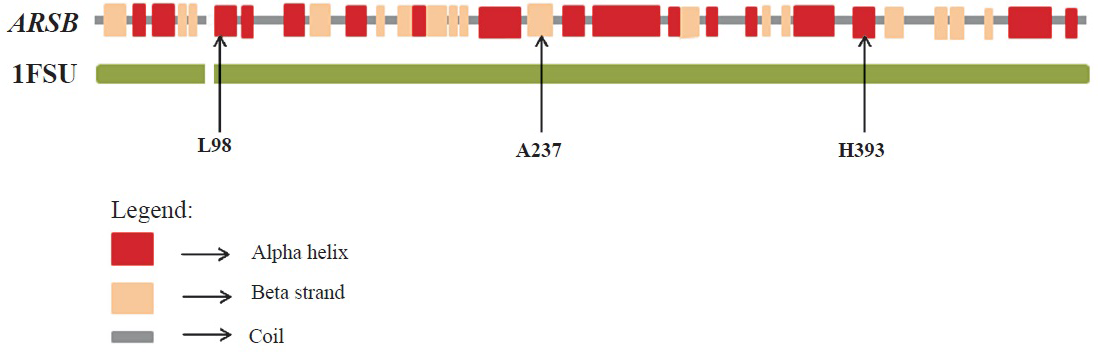
- Structural analysis of ARSB missense mutations using the 1FSU crystal structure of Protein Data Bank (PDB) database shows alteration of the protein structure of ARSB due to the missense mutations L98R, A237D and H393R.
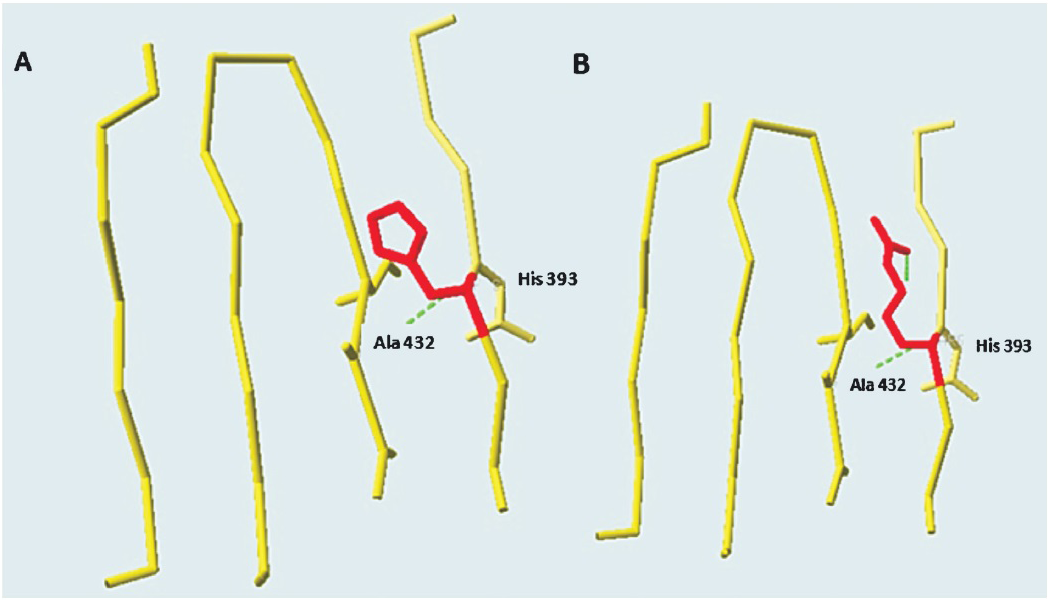
- Structural analysis of ARSB missense mutations done using SwissPdb Viewer 4.0.4. The wild type His residue at position 393 Panel (A) has been mutated to Arg Panel (B). The hydrogen bonds are shown using green dashes.
Discussion
Fifteen patients (from 14 different families) with MPS VI showed 12 different mutations. Pedigree analysis revealed that there was parental consanguinity in 11 patients from 10 different families. The patients exhibited a wide spectrum of clinical features leading to a great phenotypic variability. This confirmed the broad spectrum of genetic heterogeneity previously reported in patients with MPS VI26. We identified a wide variety of mutations in the MPS VI patients (Fig. 4, Table II). A total of nine novel mutations were identified of which five were missense mutations, three were deletions and one was a nonsense mutation. The remaining three identified mutations were known mutations42728, which are listed in the Human Genome Mutation Database29 (www.hgmd.cf.ac.uk).
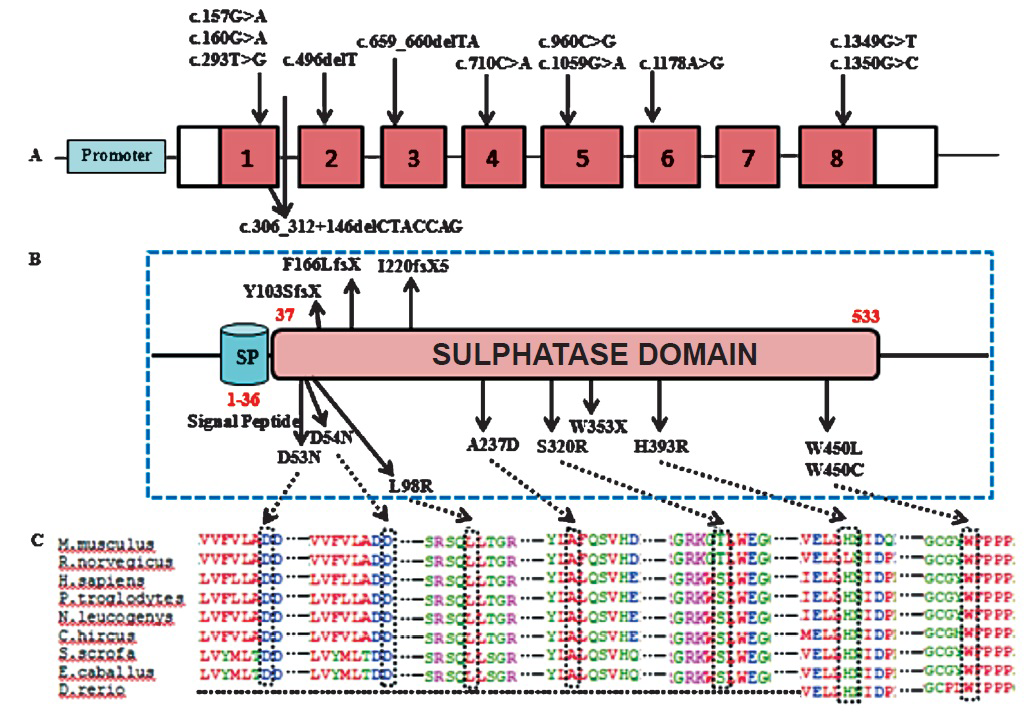
- Picture depicting (A) Mutational spectrum in ARSB; (B) Active sulphatase domain of ARSB protein; (C) Evolutionary conservation of amino acid residues in homologous ARSB proteins at the missense mutation sites.
Previous studies have documented more than 130 ARSB gene mutations worldwide so far263031. Haplotype analysis done for the mutation revealed that p.W450C may be a founder mutation in the Indian population. Extensive sequence and structural analysis has shown that the mutations D53N, D54N and A237D affect the active site of the enzyme and, therefore, may result in a severe phenotype32. The mutations identified in the patients 1 and 2 viz. p.D53N, and p.D54N, respectively resulted in a negatively charged Aspartic acid being substituted by a polar uncharged amino acid Asparagine leading to a significant alteration in the hydrogen bonding pattern in the ARSB protein. A previous report has also confirmed that p.D54N is a disease causing mutation4. Protein structural analysis suggested that the mutations L98R and H393R destabilized the protein structural core and that the three deletion mutations (c.496delT, c.306_312delCTACCAG+146del and c.659_660delTA) affected the C-terminal domain of ARSB protein3334. Earlier literature also supports that amino acid changes in the Leu98 and His393 positions are likely to be disease causing435. The reason behind the pathogenic effect of W450C and W450L mutations was not obvious from the structural analysis though the residue was found to be evolutionarily highly conserved.
In the present study, mild intellectual disability and developmental delay was found in one patient who was homozygous for the mutation p.D53N; this patient also showed a severe phenotype. The association of mental retardation with the p.D53N mutation is yet to be understood through functional studies.
MPS VI exhibits significant genetic heterogeneity, which in turn facilitates better understanding of the phenotypic variability36. Phenotypic variability includes variations in the age of onset of the disease, rate of disease progression and variation in the degree of severity of the disease. MPS VI can present with varying phenotypes ranging from a rapidly progressing (severe phenotype) to a very slowly progressing course (mild phenotype)3. In our study 12 patients had a severe phenotype, all of them manifesting disease symptoms by two years of age and then progressing to develop coarse facies, stunted growth, corneal clouding, joint contractures, hepatosplenomegaly, dysostosis multiplex and cardiopulmonary complications. One patient had an intermediate phenotype with onset of skeletal manifestations with mild facial coarsening and corneal clouding by around four years of age. Two were siblings had a mild phenotype; the elder sibling, a 11 year old girl, had mild growth retardation, corneal haziness, mild joint stiffness and early dysostosis multiplex changes and the younger sibling, a 6.5 yr old boy, had mild corneal haziness with mild joint stiffness and early dysostosis multiplex changes, without growth retardation.
Several exonic polymorphisms in the ARSB gene including p.V358M (rs1065757), p.V376M (rs1071598), p.E461E, p.P397P (rs25413) and p.S384N (rs25414) which have been previously reported43738 were also found in our study. Two intronic polymorphisms were also found more commonly amongst our patients: nine of the 15 patients had the rs25415 (chr5:78135276T>G) polymorphism and six patients had the rs6870443 (chr5:78251347A>G) polymorphism.
The establishment of genotype-phenotype association by means of theoretical and experimental techniques helps in providing better genetic counselling and more accurate prognostication. In the present study, the mutation p.D53N was found to be associated with a severe phenotype as well as with intellectual impairment. All those patients having mutations in exon 1 showed severe phenotypes. Mutations in exon 8 of the ARSB gene (p.W450C and p.W450L) were likely to be deleterious, but with varied disease progression. The mutation p.W450C in two patients resulted in a severe phenotype whereas the mutation p.W450L in two siblings resulted in a mild phenotype. The genetic and molecular bases underlying these mutations occurring at the same position but with varied severities are not yet understood and characterized completely and need to be investigated further.
Further studies involving more patients with MPS VI may lead to the identification of common and recurrent ARSB gene mutations in the Indian population, which would help in designing cost-effective testing strategies for affected families. Limitations of the study included less number of patients and unavailability of clinical details for all patients. This study also provides some insight into the genotype-phenotype association in MPS VI and further studies would facilitate better understanding of the molecular pathophysiology of MPS VI.
Acknowledgment
The authors thank the patients and their families who volunteered in the study, for their cooperation. The authors acknowledge the support provided by the Centre for DNA Fingerprinting and Diagnostics, Hyderabad, and the Indian Council of Medical Research (ICMR), New Delhi, for financial support. Dr Shubha Phadke acknowledges ICMR for funding DNA banking of the patients seen in the Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow. The first author (AU) acknowledges the Council of Scientific and Industrial Research, New Delhi, for providing Junior and Senior Research Fellowships.
Conflicts of Interest: None.
References
- The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, eds. The metabolic and molecular basis of inherited disease. New York: McGraw-Hill; 2001. p. :3421-52.
- [Google Scholar]
- Expert recommendations for the laboratory diagnosis of MPS VI. Mol Genet Metab. 2012;106:73-82.
- [Google Scholar]
- Mutational analysis of 105 mucopolysaccharidosis type VI patients. Hum Mutat. 2007;28:897-903.
- [Google Scholar]
- Differential assay of arylsulfatase A and B activities: a sensitive method for cultured human cells. Anal Biochem. 1981;117:382-9.
- [Google Scholar]
- A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215.
- [Google Scholar]
- Purification of nucleic acids by extraction with phenol:chloroform. CSH Protoc 2006. 2006;1:pii. pd.fprot4455
- [Google Scholar]
- Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289-91.
- [Google Scholar]
- Recommendations for a nomenclature system for a human gene mutations.Nomenclature Working Group. Hum Mutat. 1998;11:1-3.
- [Google Scholar]
- Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7-12.
- [Google Scholar]
- MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361-2.
- [Google Scholar]
- Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011;39:9-20.
- [Google Scholar]
- Hansa: an automated method for discriminating disease and neutral human nsSNPs. Hum Mutat. 2012;33:332-7.
- [Google Scholar]
- Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073-81.
- [Google Scholar]
- SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812-4.
- [Google Scholar]
- Haplotype analysis in population genetics and association studies. Pharmacogenomics. 2003;4:171-8.
- [Google Scholar]
- Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497-500.
- [Google Scholar]
- EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276-7.
- [Google Scholar]
- JOY: protein sequence-structure representation and analysis. Bioinformatics. 1998;14:617-23.
- [Google Scholar]
- Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol. 2002;12:14-20.
- [Google Scholar]
- SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714-23.
- [Google Scholar]
- Identification of the molecular defects in Spanish and Argentinian mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) patients, including 9 novel mutations. Mol Genet Metab. 2007;92:122-30.
- [Google Scholar]
- Identification of mutations in the arylsulfatase B gene in Russian mucopolysaccharidosis type VI patients. Genetika. 2000;36:837-43.
- [Google Scholar]
- Mucopolysaccharidosis type VI phenotypes-genotypes and antibody response to galsulfase. Orphanet J Rare Dis. 2013;8:51.
- [Google Scholar]
- The Human Gene Mutation Database: providing a comprehensive central mutation database for molecular diagnostics and personalized genomics. Hum Genomics. 2009;4:69-72.
- [Google Scholar]
- Molecular analysis of mucopolysaccharidosis type VI in Poland, Belarus, Lithuania and Estonia. Mol Genet Metab. 2012;105:237-43.
- [Google Scholar]
- Genetic studies in a cluster of mucopolysaccharidosis type VI patients in Northeast Brazil. Mol Genet Metab. 2011;104:603-7.
- [Google Scholar]
- Structure of the human arylsulfatase B gene. Biol Chem Hoppe Seyler. 1993;374:327-35.
- [Google Scholar]
- Non-sense mediated mRNA decay: terminating erroneous gene expression. Curr Opin Cell Biol. 2004;16:293-9.
- [Google Scholar]
- Pharmacological read-through of nonsense ARSB mutations as a potential therapeutic approach for mucopolysaccharidosis VI. J Inherit Metab Dis. 2013;36:363-71.
- [Google Scholar]
- Identification, expression, and biochemical characterization of N-acetylgalactosamine-4-sulfatase mutations and relationship with clinical phenotype in MPS-VI patients. Am J Hum Genet. 1996;58:1127-34.
- [Google Scholar]
- Four novel mutant alleles of the arylsulfatase B gene in two patients with intermediate form of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) Hum Genet. 1994;93:259-64.
- [Google Scholar]
- Molecular characterization of 355 mucopolysaccharidosis patients reveals 104 novel mutations. J Inherit Metab Dis. 2013;36:179-87.
- [Google Scholar]
- Maroteaux-Lamy syndrome: functional characterization of pathogenic mutations and polymorphisms in the arylsulfatase B gene. Mol Genet Metab. 2008;94:305-12.
- [Google Scholar]




