
Translate this page into:
Identification of a case of SRD5A3-congenital disorder of glycosylation (CDG1Q) by exome sequencing
*For correspondence: e-mail: neerja17aiims@gmail.com
-
Received: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Sir,
Congenital disorders of glycosylation (CDG) are the groups of disorders that involve multi-organ disease with neurological involvement due to defect in protein glycosylation. type I CDG involves defective assembly of dolichol-linked oligosaccharides, whereas type II indicates a disorder of processing of glycoproteins in the Golgi system1. Over the past decade, with the advent of next-generation sequencing and improved understanding, the number of diagnosed glycosylation disorders has risen to about one hundred2. SRD5A3-CDG (CDG1Q; MIM 612379) has been a recently described steroid 5a-reductase type 3 defects with broad clinical phenotype. Till date, only 20 patients from 14 families have been described worldwide3456789. We describe here an Indian case of SRD5A3-CDG identified by exome sequencing.
A young gravida five Hindu woman married non-consanguinously approached the Genetic Clinic, division of Genetics, department of Paediatrics, All India Institute of Medical Sciences, New Delhi, in 2014 at six weeks of gestation for genetic counselling for having a previous child with developmental delay. There was a history of two medical terminations of pregnancies for prominent cisterna magna and positive IgM for rubella, respectively. She had a prior history of one spontaneous abortion (Fig. 1A). †Proband was born at full term by forceps delivery at a birth weight of 3.75 kg and had no history of birth asphyxia. Parents noticed developmental delay at the age of five and a half months for poor head control. He had severe global developmental delay and was noticed to have impaired vision at 10 months. He developed seizures at three and half years, had delayed speech with only one to two meaningful words at the age of four years. At four years he had a weight of 18.5 kg [at +1 standard deviation (SD) WHO charts10], height of 106 cm (0 to +1 SD) and head circumference of 49 cm (at −1 SD). Examination showed the presence of mild facial coarseness, slightly deep set eyes, round nasal tip, full cheeks and thick lips (Fig. 1B); nystagmus, inverted nipples (Fig. 1C) and central hypotonia. There was no ichthyosis, deafness, hand stereotypes or ataxia. Fundus examination showed the presence of pigmentary retinopathy and mild disc pallor. There was no ocular coloboma. Visual-evoked potential showed an extinguished response. His routine haematological, renal and liver function tests were normal. Basic metabolic screening, arterial lactate and blood ammonia were normal. Neuroimaging showed mild white matter paucity without any evidence of cerebellar or vermian hypoplasia. Electroencephalogram and brainstem-evoked potential were normal. His karyotype and chromosomal microarray (CMA) were normal. CMA was performed using both copy number changes (CNCs) and single nucleotide polymorphism (SNP) probes on a whole genome array (Illumina, HumanCytoSNP-12 array)1112. The genome-wide functional resolution of this array was approximately 30 kb for CNCs and with higher resolution for all targeted regions of known cytogenetic importance. GenomeStudio-Illumina and KaryoStudio-Illumina softwares (USA) were used to analyze the data. NCBI Human Genome build 37.1, hg19 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.13/) was used as reference. In view of the presence of mild facial coarseness, inverted nipples, hypotonia and pigmentary retinopathy, the patient's blood was tested for serum isoelectric focussing along with exome sequencing.
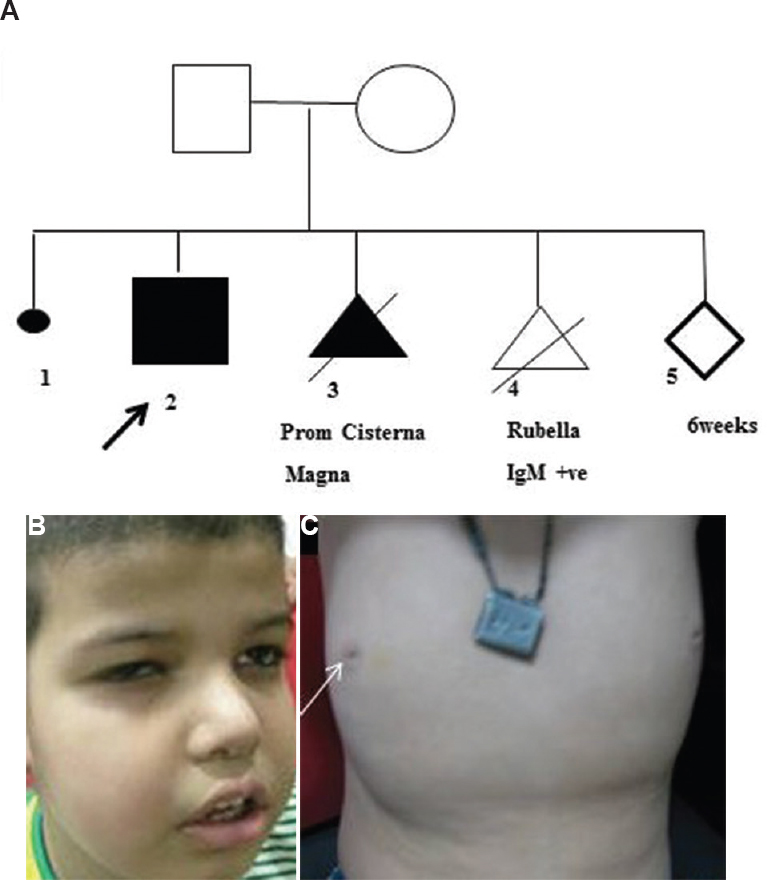
- (A) The pedigree with a medical termination of pregnancies (II-3 and II-4) in view of prominent cisterna magna. (B) The facial coarseness, slightly deep set eyes, round nasal tip, full cheeks and thick lips. (C) Inverted nipples (arrow).
The genomic DNA was sequenced using TruSight Inherited Disease Panel (Illumina, San Diego, CA, USA) that contains 552 genes associated with severe recessively inherited Mendelian diseases. The input DNA was first converted into the adaptor tagged index using Nextera DNA library preparation protocol (Illumina, San Diego, CA, USA). Target library was amplified using limited cycles PCR (ABI9700, Life Technologies, USA) steps and sequenced using the MiSeq Platform (Illumina, San Diego, CA, USA) according to the manufacturer's instructions. The trimmed FASTQ files were generated using the MiSeq Reporter from Illumina. The reads were aligned against the whole genome build hg19 using STRAND NGS v1.6 (http://www.strand-ngs.com/) which is an integrated platform that provides analysis, management and visualization tools for NGS data. The data analysis and interpretation were performed using Strand NGS v1.6 and StrandOmics (http://www.strandls.com/strandomics/; a proprietary clinical genomics interpretation and reporting platform from Strand Life Sciences)13. The variants identified were classified according to the American College of Medical Genetics and Genomics (ACMG) recommendation for standards for interpretation and reporting of sequence variations14. Exome sequencing identified the presence of a previously reported homozygous pathogenic variant c.57G>A (p. Trp19Ter) in the SRD5A3 gene (Fig. 2A-C). The variant introduces a premature termination codon and is predicted to result in a nonsense-mediated mRNA decay of the aberrant transcript15. The mutation was confirmed by Sanger sequencing. Both the parents were heterozygous for the same mutation. Prenatal diagnosis at 16 wk showed the foetus as unaffected. Serum transferrin isoelectric focussing showed a characteristic type I pattern with elevated asialotransferrin of 12.93 per cent (reference range-1.01-4.71%), disialotransferrin of 20.21 per cent (reference range -9.15-16.74 %) with low tetrasialotransferrin of 21.58 per cent (reference range -29.32-44.51%). The presence of disialotransferrin and a decreased ratio of trisialotransferrin to disialotransferrin of 0.78 (normal ratio >1.25) was supportive of a type I pattern. As glycosylation is important for the insulin-like growth factor (IGF) system16, the proband's serum IGF-binding protein 3 (IGFBP3) levels were analysed which were found to be normal. Till the last follow up seizures are controlled with sodium valproate, but he had severe developmental delay and was undergoing occupational therapy. The permission to report this case was obtained from the institutional ethics committee and written informed consent was obtained from the parent.
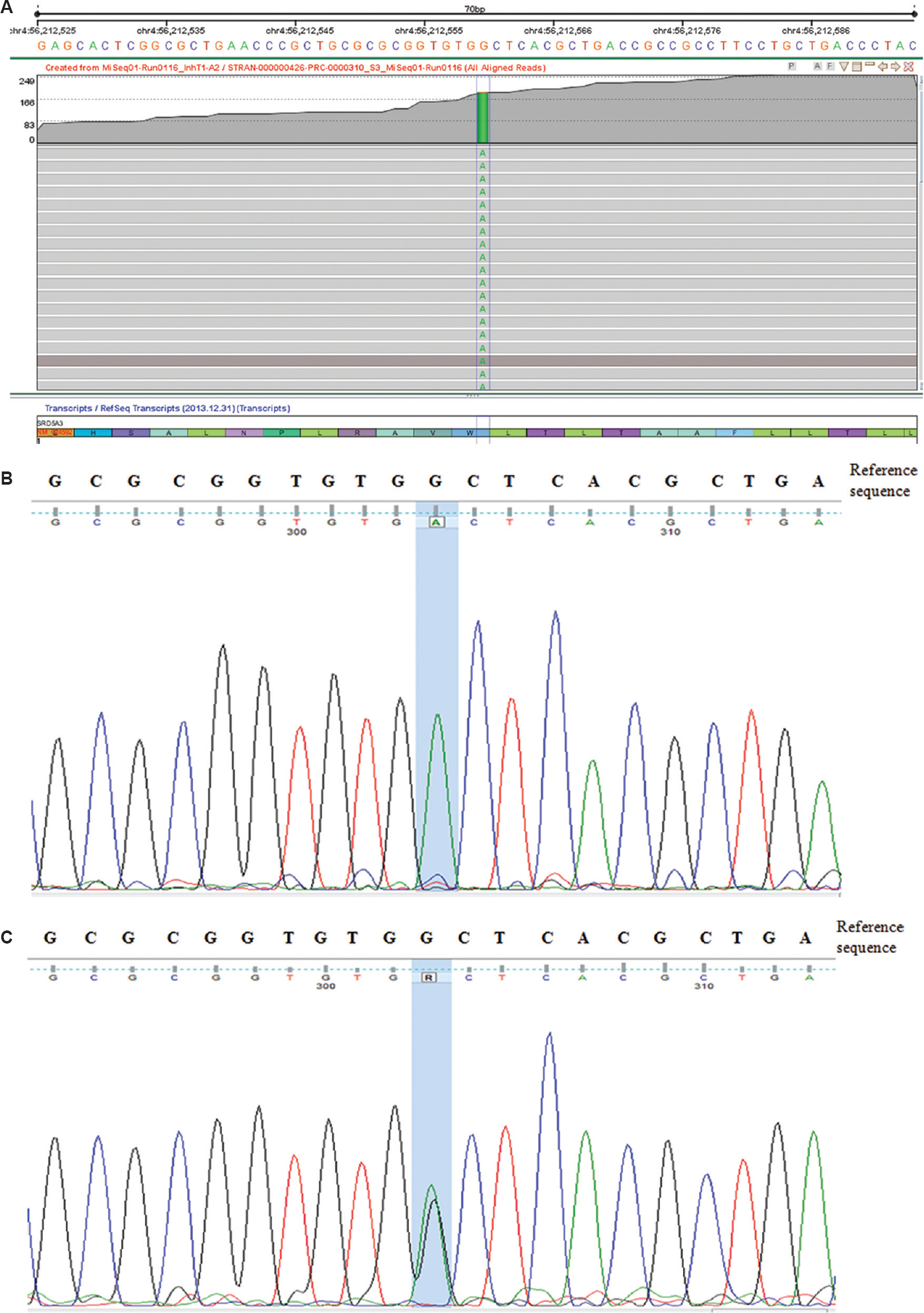
- (A) Genome browser view in Strand NGS showing the homozygous change c.57G<A (p.Trp19Ter) in the SRD5A3 gene. (B) Sanger sequencing data from the proband showing the homozygous nucleotide change ‘G<A’ (c.57G<A) in the SRD5A3 gene. (C) Sanger sequencing data from the parents and the foetus showing the heterozygous nucleotide change ‘G<A’ (c.57G<A) in the SRD5A3 gene, suggesting their carrier status.
SRD5A3-CDG is an uncommon disorder of glycosylation and is characterized by intellectual disability, visual impairment, nystagmus, muscular hypotonia and ataxia in the majority12. About half of these patients can have associated cerebellar or vermian hypoplasia. Other features include microcytic anaemia, ichthyosiform dermatosis, abnormal liver enzymes and coagulation abnormalities. SRD5A3 gene at 4q12 locus encodes for a protein that belongs to the steroid 5-alpha reductase family and polyprenol reductase subfamily. It is involved in the production of androgen 5-alphadihydrotestosterone from testosterone and is also necessary for the conversion of polyprenol into dolichol, which is an essential step for N-linked glycosylation of proteins4. SRD5A3 is highly expressed in brain, especially the hippocampus and cerebellum, and is important for brain development. Truncating mutations in SRD5A3 gene hampers protein glycosylation and causes two phenotypically distinct allelic disorders SRD5A3-CDG and Kahrizi syndrome17. Twenty cases from 14 families with several truncating mutations in this gene have been described with SRD5A3-CDG3456789. Of the previously described cases4579, five patients (four unrelated families) had homozygous p. Trp19Ter mutations. Of these, three families were Turkish and one was from Pakistani and all had consanguinity.
The presence of homozygous mutation in an apparently non-consanguineous couple is not an uncommon finding. In spite of a high genetic diversity due to the evolutionary history of the Indian ethnic groups, it is governed by a high degree of endogamy practices based on the religion, language and geographical location, thereby resulting in a high prevalence of recessive genetic disorders1819. In the present case, though there was no apparent consanguinity within the immediate family members, it could be possible that there was a lack of awareness of the likelihood of a marriage between distant relatives several generations before.
Our patient showed mild white matter paucity that may be age related. In addition, our patient had facial coarseness and inverted nipples, not described in previously reported patients. He did not have short stature and had normal serum IGFBP3 in contrast to previously described cases9. Others have shown that although both inter- and intra-familial variability occurs, but no specific genotype, phenotype correlation has been observed in these patients57.
In the present study, there was a delay of about four years for the diagnosis of the patient and he underwent a battery of tests without any confirmatory diagnosis until the age of five years. This delay was probably due to the evolution of phenotype over a period of time and non-availability of the exome analysis earlier. The presence of pigmentary retinopathy, hypotonia and inverted nipple raised the suspicion of the CDG. In view of pregnancy, both targeted exome and serum isoelectric focussing was performed almost simultaneously on the proband's blood. The presence of previously described homozygous pathogenic variant c.57G>A (p. Trp19Ter) in SRD5A3 gene on exome analysis and a phenotypic and biochemical correlation of the findings helped in making a conclusive diagnosis and facilitated timely prenatal diagnosis.
Our study highlights the importance of follow up evaluation of a patient with intellectual disability, validation of exome results with further clinical phenotyping and biochemical and/or functional studies.
Acknowledgment
Authors thank the family for their cooperation in this study, and also acknowledge Dr Anil Jalan for performing isoelectric focussing.
Financial support & sponsorship: None.
Conflicts of Interest: None.
References
- Other forms of congenital disorder of glycosylation. In: Atlas of inherited metabolic disease (3rd ed). London: CRC Press; 2011.
- [Google Scholar]
- Congenital disorders of glycosylation: A concise chart of glycocalyx dysfunction. Trends Biochem Sci. 2015;40:377-84.
- [Google Scholar]
- A new subtype of a congenital disorder of glycosylation (CDG) with mild clinical manifestations. Neuropediatrics. 2001;32:313-8.
- [Google Scholar]
- Life with too much polyprenol: Polyprenol reductase deficiency. Mol Genet Metab. 2012;105:642-51.
- [Google Scholar]
- Adult phenotype and further phenotypic variability in SRD5A3-CDG. BMC Med Genet. 2014;15:10.
- [Google Scholar]
- A novel cerebello-ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism. Brain. 2010;133:3210-20.
- [Google Scholar]
- A new case of CDG-x with stereotyped dystonic hand movements and optic atrophy. J Inherit Metab Dis. 2002;25:126-30.
- [Google Scholar]
- Phenotypic expansion of congenital disorder of glycosylation due to SRD5A3 null mutation. JIMD Rep. 2016;26:7-12.
- [Google Scholar]
- WHO Child Growth Standards based on length/height, weight and age. Acta Paediatr. 2006;450:76-85.
- [Google Scholar]
- High-resolution genomic profiling of chromosomal aberrations using infinium whole-genome genotyping. Genome Res. 2006;16:1136-48.
- [Google Scholar]
- Application of SNP array for rapid prenatal diagnosis: Implementation, genetic counselling and diagnostic flow. Eur J Hum Genet. 2011;19:1230-7.
- [Google Scholar]
- Detection of high frequency of mutations in a breast and/or ovarian cancer cohort: Implications of embracing a multi-gene panel in molecular diagnosis in India. J Hum Genet. 2016;61:515-22.
- [Google Scholar]
- ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294-300.
- [Google Scholar]
- Nonsense-mediated mRNA decay: Splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004;5:89-99.
- [Google Scholar]
- IGF system in children with congenital disorders of glycosylation. Clin Endocrinol (Oxf). 2009;70:892-7.
- [Google Scholar]
- An autosomal recessive syndrome of severe mental retardation, cataract, coloboma and kyphosis maps to the pericentromeric region of chromosome 4. Eur J Hum Genet. 2009;17:125-8.
- [Google Scholar]
- Complex genetic origin of Indian populations and its implications. J Biosci. 2012;37:911-9.
- [Google Scholar]
- Unity in diversity: An overview of the genomic anthropology of India. Ann Hum Biol. 2014;41:287-99.
- [Google Scholar]




