
Translate this page into:
Gene expression & biochemical analysis in alkaptonuria caused by a founder pathogenic variant across different age groups from India
For correspondence: Prof. Sumita Danda, Department of Clinical Genetics, Christian Medical College, Vellore, Tamil Nadu 632 004, India e-mail: sumita_danda@hotmail.com
-
Received: ,
Accepted: ,
Abstract
Background & objectives
Alkaptonuria (AKU) is an autosomal recessive disease wherein biallelic pathogenic variants in the homogentisate 1,2- dioxygenase (HGD) gene encoding the enzyme homogentisate 1,2 dioxygenase cause high levels of homogentisic acid (HGA) to circulate within the body leading to its deposition in connective tissues and excretion in urine. A homozygous splice donor variant (c.87+1G>A) has been identified to be the founder variant causing alkaptonuria among Narikuravars, a group of gypsies settled in Tamil Nadu.
Methods
Blood and urine samples of 30 homozygous splice site donor variant individuals (2 groups aged 7-20 and 21-83 yr, with 9 and 21 individuals, respectively), carriers and 30 wild-type individuals from the Narikuravars were collected during field visits after obtaining informed consent. Clinical evaluation and genetic counselling were done. The plasma and urine HGA levels were estimated by high-performance liquid chromatography. RNA was extracted from the peripheral blood and reverse transcribed. Sanger sequencing was done to check the consequence of the splice donor variant. Relative quantification of the cDNA in the three groups was done by real-time qPCR (RT-qPCR) studies using reference genes followed by Pearson’s correlation analysis.
Results
In our cohort, among the affected alkaptonuria individuals, the minimum age for eye pigmentation detected was 23 yr. Similarly, the minimum age for back pain and any joint pain was 30 yr and 38 yr, respectively. Sequencing of the cDNA confirmed exon 2 skipping in affected individuals. In comparison to the normal individuals, the affected individuals showed reduced HGD expression. HGD relative expression showed a significant correlation (P<0.05) with mean plasma HGA levels in the younger (≤22 yr) age group but not in the older one. There was also a significant correlation (P<0.05) of reduced HGD expression with back pain in the 21-37 yr age group. Increasing age showed a positive correlation with circulating mean plasma HGA levels and a negative correlation with excreted HGA.
Interpretation & conclusions
As per the authors’ knowledge, this is the first study to confirm the functional effect by RT-PCR of this highly prevalent founder HGD variant causing alkaptonuria in the Narikuravar community. Both plasma and urinary HGA levels correlated well with the gene expression of this variant and could serve as potential markers of AKU severity for those with this variant.
Keywords
Biomarker
exon skipping
HGD gene
HPLC
plasma HGA
RT-qPCR
South India
splicing variant

Alkaptonuria (AKU, MIM:203500) is an autosomal recessive disorder caused due to biallelic pathogenic variants in the homogentisate 1,2-dioxygenase (HGD) gene, which leads to deficiency of the enzyme homogentisate dioxygenase that is involved in the tyrosine degradation pathway1. AKU’s three major cardinal signs are homogentisic acid (HGA) in urine, ochronosis (pigmentation of connective tissues) and osteoarthritis. The HGA that is excreted in the urine turns dark upon oxidation hence giving the name ‘black urine disease’. This generally happens from birth and in many cases remains unnoticed until the severe symptoms occur. Ochronosis in the eyes, ears, skin and arthritis in the spine and larger joints show delayed manifestations, generally after the third decade of life. Worldwide, more than 250 unique variants have been identified in the HGD to cause alkaptonuria (HGD mutation database, http://hgddatabase.cvtisr.sk/ )2. The incidence is 1 in every 250000 to one million live births, making it a rare disorder3.
In India, there are many case reports of AKU4-7 and it was first reported among the Indian Narikuravars by Jebaraj et al8 in 2005. A homozygous splice site variant (NM_000187.4:c.87+1G>A) was identified to cause AKU among this community9. The founder effect of this variant was further confirmed by Danda et al10. The study was conducted in seven different districts of the State Tamil Nadu, and a prevalence of AKU was found to be 8.4 per cent in the gypsy population of south India10.
This present study was aimed to demonstrate by RT-PCR the functional effect of the splice donor variant (c.87+1G>A) on HGD gene expression in genetically tested homozygous individuals (c.87+1G>A/c.87+1G>A) at different stages of the disease and compare them with age-matched wild-type controls (wt/wt) and heterozygotes (c.87+1G>A/wt) from the same Narikuravars community. Additionally, biochemically, the plasma and urine HGA levels were estimated among the subsets to correlate with gene expression studies.
Materials & Methods
This study was conducted at the department of Clinical Genetics, Christian Medical College (CMC), Vellore, Tamil Nadu, India. This study was approved by the Institutional Review Board and Ethics Committee of CMC, Vellore.
Study participant recruitment
Field visits to eight different Narikuravar settlement areas in six different districts of Tamil Nadu, namely Vellore, Kanmangalam, Trichy, Kanchipuram, Chennai, Perambalur, and Salem. Study participants, aged 7-83 yr, were recruited after obtaining verbal and written consent/assent. Detailed clinical evaluation and genetic counselling were also done. Symptomatic and asymptomatic affected individuals were verified by checking for dark colour change of urine upon the addition of sodium hydroxide. The family members of the affected individuals whose urine did not change color were recruited in the heterozygotes or wild-type control groups and were further segregated by Sanger DNA sequencing11. Pregnant women were excluded from the study. The sample size was 30 individuals homozygous for c.87+1G>A, 25 heterozygous carriers (c.87+1G>A/wt) and 32 wildtype controls. The study design is provided in Supplementary figure 1.
Clinical features of affected individuals
The details of gender, age at the time of recruitment, family history, consanguinity, dark colour change of urine, presence of pigmentation in the sclera of eyes, ear helices, the skin of the hands, presence of pain in the back and weight-bearing joints such as shoulder, elbow, hip, knee and ankle and presence of other co-morbidities were noted in the proforma.
Sample collection
About 10 ml peripheral blood was collected in heparin, EDTA and PAX, blood RNA tubes (PreAnalytiX by Qiagen) to extract plasma, DNA and RNA, respectively. PreAnalytiX instructions of PAX blood RNA tube were followed during blood collection. Heparin blood was processed within 2h of sample collection for plasma separation.
Urine was collected in urine collection containers. For HGA estimation, 4 ml of urine was added to falcon tubes containing 100 µL of 0.1 M hydrochloric acid (HCl). All the samples were stored in ice during transit to Molecular Genetics Laboratory, Department of Clinical Genetics, CMC, Vellore.
Variant screening by Sanger DNA sequencing
DNA was extracted from the EDTA blood using Qiagen’s DNA Mini kit. The quality and quantity of the DNA were checked using the Nanodrop Spectrophotometer (Thermo Fischer Scientific). PCR amplification of HGD exon 2 (NM_000187.4; human genome version GRCh38) was performed using specific primers followed by Sanger sequencing to check presence/absence of the splice donor variant.
Estimation of HGA levels in urine and plasma
For all affected individuals, HGA levels in plasma and urine were quantified by High-Performance Liquid Chromatography (HPLC) at the departments of Clinical Biochemistry and Clinical Pharmacology, CMC, Vellore, respectively10. Briefly, according to the Bory method12, the heparinized blood was centrifuged at 1500g, 10 minutes, 4°C and to every 100 µl of supernatant, 10 µl 5.8M perchloric acid was added. A second centrifugation was performed and the supernatant removed. The sample was stored at -20°C.
RNA isolation and reverse transcription
RNA was isolated using a PAX Blood RNA kit as per the manufacturer’s instructions (PreAnalytiX GmbH, Switzerland). The quality and quantity of the RNA was checked in the Nanodrop Spectrophotometer. The integrity of the RNA was checked based on the appearance of 2 bands (1 band for 28S and another 18S rRNA) after gel electrophoresis using 1% agarose. The RNA was then converted to cDNA using Qiagen’s Quantinova reverse transcription kit as mentioned in Supplementary Table I.
RT-PCR and cDNA sequencing
Primers were designed using Premier BioSoft’s Beacon Designer™ free edition software (https://www.premierbiosoft.com/qpcr/) and UCSC insilico PCR tool (https://genome.ucsc.edu/cgi-bin/hgPcr) based on the reference transcript HGD (NM_000187.4; human genome version GRCh38). PCR amplification of RT product specific to HGD exons 1-3 was performed, followed by gel electrophoresis. Sanger sequencing of the cDNA was done for selected samples to identify the changes in splicing events using in-house facilities at CMC Vellore.
Real-time qPCR
Based on the minimum information necessary for publication of RT-qPCR experiments (MIQE guidelines)13-15, seven reference genes were chosen from previous experiments carried out using peripheral blood (Supplementary Table II): Glyceraldehyde-3-phosphate dehydrogenase (GAPDH - 115 bp)16, β-actin (ACTB - 110 bp)16, 18s ribosomal RNA (18srRNA - 100 bp)16, β2-Microglobulin (B2MG-138bp)16, Peptidyl isomerase A (PPIA - 141 bp)16, TATA-box binding protein (TBP - 117 bp)16 and Hypoxanthine phosphoribosyl transferase1 (HPRT1 - 94 bp)17. Out of them, the two most stable genes were identified for this study using 27 homozygous (3 were excluded from the analysis), 7 heterozygous (c.87+1G>A/wt) and 17 wild-type controls (wt/wt) representative samples. The real-time qPCR assay was optimized, and a strong linear relationship (r2 = 0.9517) between the Ct and log of target concentration was demonstrated for the HGD primers. The E value obtained was 104.98 per cent (or 2.05) (Supplementary Fig. 2). Using Genorm (Qbase+), NormFinder and BestKeeper software, the two most stable reference genes for this study were identified as HPRT1 and PPIA (Supplementary Table III).
HGD transcript levels were estimated by relative quantification using Quantinova SYBR Green qPCR Master Mix (2X) (Qiagen, Germany) in Bio-Rad CFX96 thermal cycler. Real-time PCR was performed in duplicates in a 96-well plate and run according to the manufacturer’s recommendation. For negative controls, water was added instead of the cDNA template. All the results were analyzed using the CFX Maestro software ( https://www.bio-rad.com/en-in/product/cfx-maestro-software-for-cfx-real-time-pcr-instruments?ID=OKZP7E15 ). Normalization to reference genes and the relative quantification of HGD was performed in qBase+ software18. Samples which showed variable normalization factors were considered outliers and excluded from the expression analysis. The details of the real time assay design is mentioned in Supplementary figure 3.
Statistical analysis
Data was analyzed using SPSS v21. Data are represented as mean±SE. The significance of correlations was determined using Pearson’s correlation analysis. P values <0.05 will be considered statistically significant.
Results
Demographic details
In the eight settlements visited, samples were collected from 92 related individuals. Among them, 30 individuals were screened positive by testing the urine for the dark colour change and were divided into two groups aged 7 to 20 and 21 to 83 yr, with 9 and 21 individuals, respectively. All the 30 individuals harboured the splice donor variant c.87+1G>A in homozygous state. Among the unaffected individuals, 25 were heterozygous carriers (c.87+1G>A/wt) and 32 were wild-type for the variant. The mean age was 37 yr in the homozygous and wildtype groups and 35 yr in heterozygous (c.87+1G>A/wt) group. The percentage of males were 40, 52 and 44 per cent in the homozygous, heterozygous (c.87+1G>A/wt) and wildtype groups, respectively.
Clinical features of affected individuals
All the homozygous individuals showed a dark colour change in their urine. Among them, the minimum age at which pigmentation (in the eye) was observed was a 23 yr old male. The minimum age for back pain to appear was 30 yr. The minimum age at which pain started in any of the weight-bearing joints (hip, knees, ankles or shoulders) was 38 yr (Table I). Based on this, the affected individuals were subdivided into age groups of 0-20 yr, 21-37 yr, and 38-85 yr for further analysis.
| Clinical features | Frequency | Percentage (%) | Minimum age (yr) | Maximum age (yr) |
|---|---|---|---|---|
| Dark urine | 30 | 100 | 7 | 82 |
| Pigmentation | 18 (27) | 66.7 | 23 | 70 |
| Abdominal pain | 6 (27) | 22.2 | ||
| Back pain | 19 (29) | 65.5 | 30 | 82 |
| Joint pain | 15 (29) | 51.7 | 38 | 81 |
| Hip joint pain | 13 (29) | 44.8 | 38 | 81 |
| Knee joint pain | 9 (28) | 32.1 | 40 | 81 |
| Ankle joint pain | 6 (27) | 22.2 | 40 | 65 |
| Shoulder joint pain | 3 (26) | 11.5 | 40 | 65 |
Homogentisic acid levels in urine and plasma samples
HPLC detected HGA in the urine and plasma of affected, homozygous individuals (Table II). Among the affected individuals, plasma HGA levels were observed to be higher than the urine HGA levels within each age group. It was observed that as age increased, mean plasma HGA levels increased (Fig. 1) and mean urine HGA levels decreased.
| n | Minimum | Maximum | Mean±SE | |
|---|---|---|---|---|
| Urine HGA value (µM) | 30 | 3.91 | 76.7 | 22.55±3.15 |
| Plasma HGA values (µM) | 30 | 9.49 | 618.5 | 83.34±19.53 |
SE, standard error
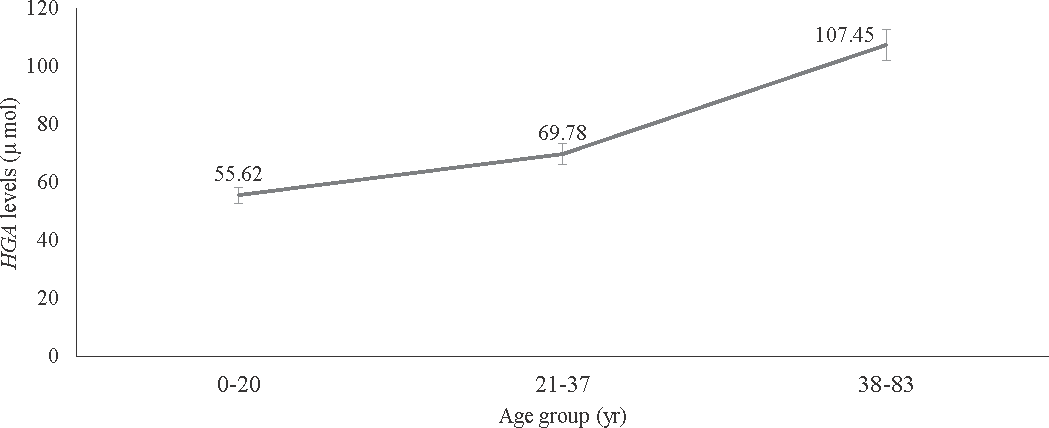
- Distribution of the mean plasma homogentisic acid (HGA) levels detected by high performance liquid chromatography (HPLC) across different age groups.
Reverse transcription and cDNA sequencing
cDNA sequencing of the homozygous, heterozygous (c.87+1G>A/wt) and wild-type (+/+) samples showed that the exon 2 was skipped as a result of the splice donor variant c.87+1G>A (Fig. 2).
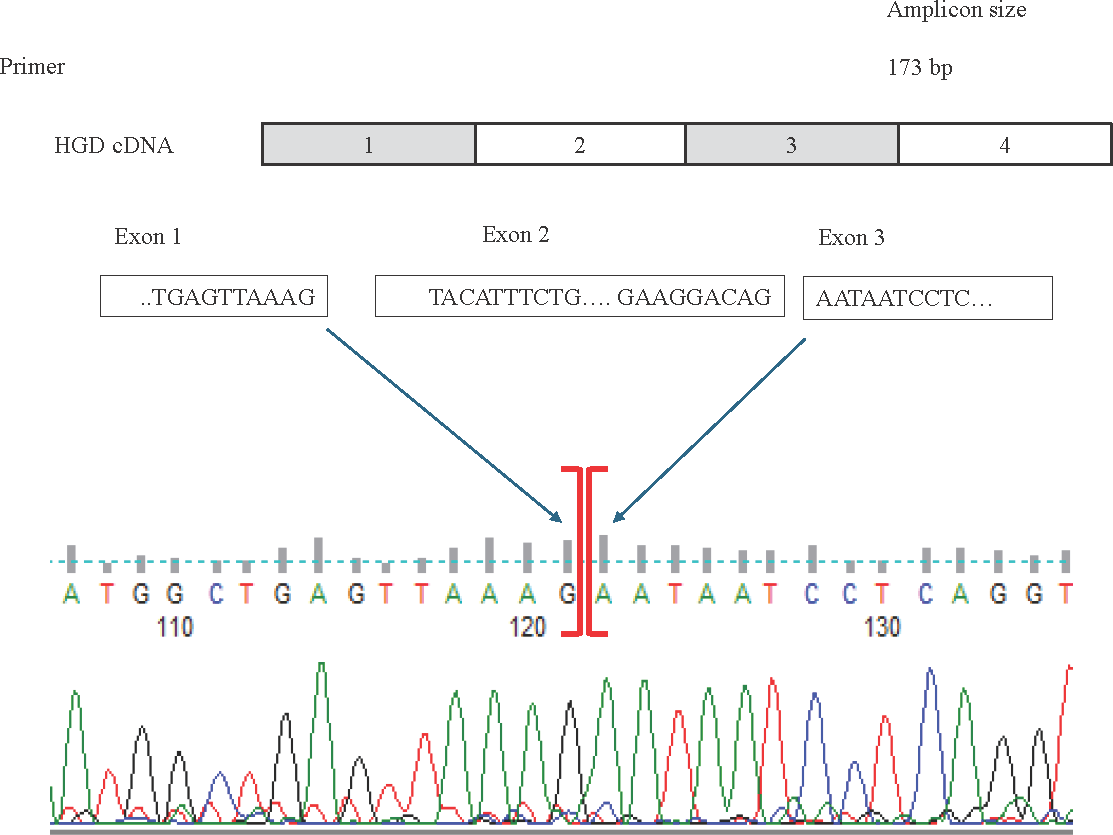
- Top: The schematic representation of the location of the forward and reverse RT-qPCR primers spanning exons 1 to 3 in the normal HGD cDNA transcript. Below: HGD cDNA sequence showing exon 2 skipping in the affected individuals carrying the splice donor variant in comparison to the heterozygous (carriers) and the normal individuals (wildtype).
HGD gene expression
All the samples that were real-time amplified along with melting curve analysis showed the homozygous (-/-), heterozygous (c.87+1G>A/wt) and wild-type (+/+) individuals having different melting peaks owing to the amplicon sizes in each case. The homozygous samples (-/-) produced a melting peak at 78°C temperature, which implies the presence of shorter amplicon. The melt peak of the wild-type samples (+/+) was at 80.50°C temperature, implying the normal amplicon. Heterozygous samples (c.87+1G>A/wt) showed two melt peaks, indicating the presence of both the normal amplicon and the short amplicon (Fig. 3A and B). This further confirms that the splice donor variant causes exon 2 skipping leading to an abnormal transcript.
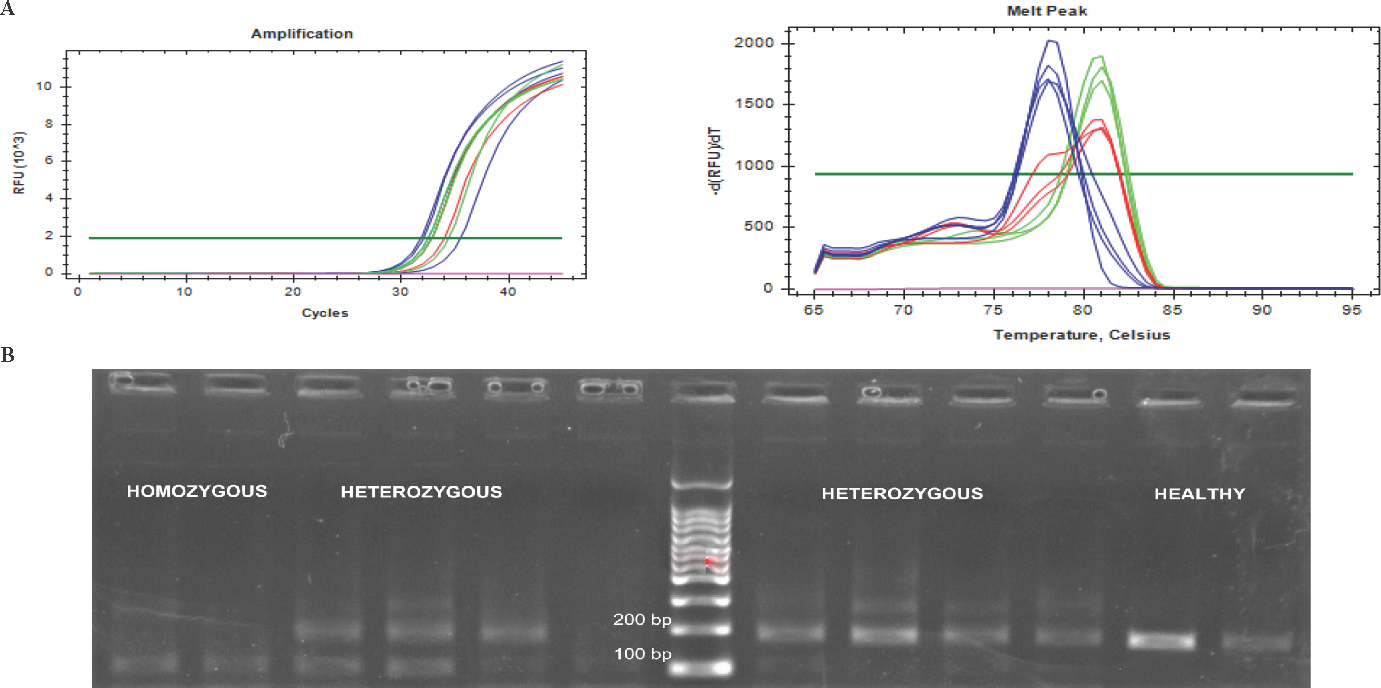
- (A) Real time qPCR (RT-qPCR) amplification plots and the melting curve peaks of the homozygous (blue), heterozygous (red) and healthy individuals (green). Note the shift in the melt peak of the homozygous samples (at 78°C) in comparison to that of healthy samples (at 80.50°C). (B) The gel electrophoresis photo showing bands at 100 bp for the homozygous samples and at 173 bp for the healthy individuals. The heterozygous samples showed bands of both sizes.
The HGD expression was quantified in relation to the two reference genes, HPRT1 and PPIA and analyzed in qbase+ software in 27 homozygous, seven heterozygous (c.87+1G>A/wt) and 17 wild-type samples after removing outliers based on variable normalization factors. The affected individuals showed overall reduced gene expression in comparison to the unaffected individuals (Fig. 4). Among the affected groups, the 0-20 yr age group (n=8) showed a comparatively higher expression than that of the other two age groups (n=6 and 13) indicating that gene expression reduced with advancing age. Pearson’s correlation analysis showed plasma HGA levels significantly correlated (P<0.05) with HGD relative expression in the 0-20 yr age group. The presence of back pain also showed significant correlation (P<0.05) with HGD relative expression in the 21-37 years age group.
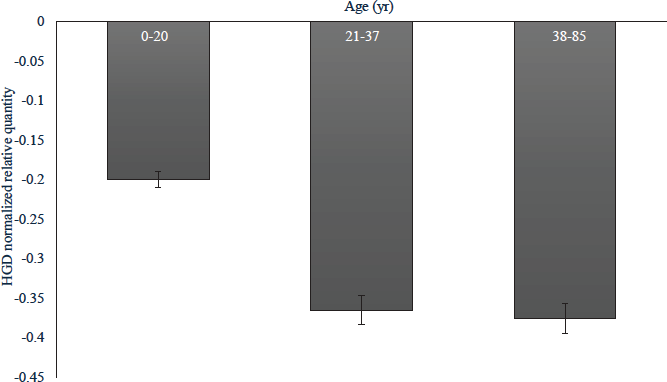
- The HGD relative gene expression in homozygous samples of different age groups.
Discussion
This study reports the reduced HGD gene expression for a founder variant by RT-PCR denoting exon 2 skipping and a functional reduction of protein leading to alkaptonuria. The gene expression of the common genotype across different age groups in correlation with the manifestation of the major cardinal signs (dark colour change of urine, ochronosis and osteoarthritis) and the HGA levels in the plasma and urine have been documented in this report. To the best of our knowledge, this is the first study which compares the gene expression changes across 27 affected (homozygous) individuals with seven heterozygous carriers and 17 wild-type individuals for a single genotype. This study is also the first of its kind to demonstrate the HGD exon 2 skipping because of the splice donor variant, c.87+1G>A, identified among affected individuals. This variant is predicted to cause the deletion of 24 amino acids coded by exon 2, namely p.(Tyr6_Gln29del). Exon skipping was confirmed by cDNA sequencing. The homozygous samples showed a shift in the melting peak and a relatively reduced gene expression when compared to the heterozygous carriers and wild-type individuals.
Another study in Lebanon reports a homozygous deletion of 649 bp, which encompasses exon 2 entirely and the flanking intronic DNA sequence19,20. They modelled the tertiary structure of the mutant protein and reported the absence of a β-pleated sheet corresponding to the missing exon 2, while the rest of the structure remained unaffected. They observed that nine novel α-helical coils substituted the eight coils of the native HGD protein19. These changes result in the deleterious enzyme leading to the manifestation of AKU symptoms. The same effect applies to the splicing variant c.87+1G>A since it caused exon 2 skipping, as confirmed by cDNA sequencing.
In India, a spectrum of variants (NM_000187.3 p.Ser59Alafs*52, p.Tyr62Cys, p.Leu116Pro, p.Gly121Glu, p.Ala122Val, p.Val157Phe, p.Glu168Asp, p.Met186Lys, p.Arg225His, p.Gly251Asp, p.Ser340Ile, p.Arg347*, p.Gly360Arg, c.775-16T>A, c.469+6T>C, p.Arg53Gln, p.Gly270Arg, c.550-2A>C, c.87+1G>A (HGD mutation database) have been identified in AKU patients2,10. Mining the Genome Asia 100K database revealed another ten variants of uncertain significance in Indians (p.Ile391Thr, p.Pro308Arg, p.Arg237HIs, p.Ile231Thr, p.Val192Ile, p.Asn139Ser, p.Lys126Asn, p.Ala105Val, p.Pro103Thr and p.Ser47Leu)21. The p.Ser47Leu variant has already been reported in several AKU patients from Slovakia, Hungary and Italy (HGD mutation database)22,23. The remaining variants are extremely rare in other ethnic groups, as per the gnomAD database, and require functional studies to resolve their impact. Interestingly the commonest variant, p.Ala122Val in the non-gypsy Indian population has been reported as founder variant in Jordan10
Mostly, the dark colour change of the urine remains unreported until chronic pain in the spine and/or joints develops. Many of the pathological changes have been studied in samples obtained from autopsy24, or post-surgical samples like heart tissue25, hip and knee joints20. Osteoclasts obtained from PBMCs26 were recently used to study bone pathophysiology.
Our study suggests reducing gene expression with advancing age which needs to be further studied as additional factors such as oxidative stress or auto-inflammation itself may regulate transcription of the mutated gene.
This present cross-sectional study in a small cohort gives a statistically significant correlation of HGD expression levels to the mean plasma HGA levels of the 0-20 yr age group. This may point to plasma HGA as a potential biomarker, which needs to be replicated in larger cohorts. A study focussing on the bone pathophysiology in AKU, reported that altered osteoclastogenesis was observed in AKU children and demonstrated that AKU adults had high levels of receptor activator of nuclear factor kappa-Β ligand (RANKL)26. Another study correlated serum levels of serum amyloid A (SAA) protein, protein thiolation index (PTI) and chitotriosidase activity to AKU severity, indicating the role of both oxidative stress and inflammation in AKU progression because of the chronic injury caused by HGA accumulation27. They also reported that even though AKU patients were symptom-free in the first decades of life, a subclinical inflammation might be present even in young asymptomatic AKU subjects, indicating a possible role of SAA as an early biomarker of disease27. Such correlations can be a potential area for future studies to determine factors responsible for variation in disease susceptibility, such as intragenic polymorphisms, and develop approaches that alleviate symptoms.
A thorough clinical investigation of this cohort using assessment tools such as the AKU severity scale index (AKUSSI)28 and quality of life predictors can help develop a timeline of how the disease progresses in severity. Metabolomic and proteomic studies to study the individual changes concerning diet, age, co-morbidities, lifestyle, environmental and other genetic factors can also give clues to the disease progression stages and identification of more precise biomarkers. Many monogenic disorders have intrafamilial variability despite having the same mutation as seen in conditions such as cystic fibrosis where modifier genes play a role in gene regulation29. Similarly, SMN2 copy number modifies the natural history and treatment outcomes in spinal muscular atrophy30. Single nucleotide polymorphisms (SNPs) have a modifying effect on a genetic disease severity or drug response31. Additional comorbidities such as obesity and diabetes mellitus can affect metabolic and biochemical parameters. This needs to be studied in AKU.
We also report that as age increases, increased accumulation of HGA takes place, and the excretion of HGA decreases, which may be influenced by renal function. This implies that plasma HGA levels can be a potentially useful marker to monitor disease severity and therapeutic response in the younger age group. As age progresses, renal involvement and impaired function may make this measurement unreliable. The role of kidneys is significant in determining HGA levels, as evidenced by the study by Ranganath et al32. However, individual variant responses may be variable. Gene expression also correlated significantly with back pain in the 21-37 yr age group.
Antisense oligonucleotide therapy, which uses a target RNA to induce protein knockdown or restoration, has shown unprecedented specificity since it allows targeting specific transcript isoforms or a specific member in a family of closely related proteins33. Moreover, since they intervene at a genetic level, they provide therapeutic options, particularly for inherited diseases such as Familial hypercholesterolemia, Duchenne muscular dystrophy, Spinal muscular atrophy types I, II and III and many others33. For the common splice donor variant causing Alkaptonuria, targeted antisense oligonucleotide therapy could be a potential intervention to correct the genotype.
The Narikuravars lead a nomadic lifestyle and remain an isolated community in different parts of Tamil Nadu, India. They do not have easy and affordable access to healthcare. Many of the affected individuals remain undiagnosed until the debilitating orthopaedic complications develop. Even if identified at an early age, they do not reveal that they excrete dark colour urine due to social discrimination and stigma. The number of alkaptonuria patients among Narikuravars is underestimated since more individuals could not be screened due to their nomadic lifestyle. The affected individuals from the general population are also being identified. This emphasizes the alarming need for a therapeutic intervention, especially in India, which can delay the onset of symptoms and thereby help in increasing the quality of life of the affected individuals.
Overall, this study evaluated 27 affected individuals with the same splice site founder variant. It reports the age of onset of the cardinal signs of AKU, the plasma and urine HGA levels and the HGD gene expression of the homozygous, heterozygous and wild-type individuals across different age groups among the Narikuravars. As per authors’ knowledge this study is also the first of its kind, to demonstrate the HGD exon 2 skipping because of the splice donor variant, c.87+1G>A identified among affected individuals. Such molecular correlations with the different stages of disease progression can help in gaining more insights into the disease mechanisms for the development and/or monitoring of therapeutic interventions. The role of food, environment and other genetic factors needs further assessment to answer the phenotypic variability observed despite the single genotype.
Acknowledgment
Authors acknowledge the technical support and guidance of Dr Isaac Jebaraj, Christian Medical College, Vellore.
Financial support & sponsorship
This work was supported by the grant (reference no. SR/WOS-A/LS-72/2017), received from Department of Science & Technology, Government of India under Women Scientist Scheme to first author SSCA to carry out this work.
Conflicts of Interest
None.
Use of Artificial Intelligence (AI)-Assisted Technology for manuscript preparation
The authors confirm that there was no use of AI-assisted technology for assisting in the writing of the manuscript and no images were manipulated using AI.
References
- The incidence of alkaptonuria: A study in chemical individuality. 1902. Mol Med. 1996;2:274-82.
- [PubMed] [PubMed Central] [Google Scholar]
- Identification of 11 novel homogentisate1,2 dioxygenase variants in alkaptonuria patients and establishment of a novel LOVD-based HGD mutation database. JIMD Rep. 2012;4:55-65.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Alkaptonuria: Current perspectives. Appl Clin Genet. 2020;13:37-47.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Alkaptonuria: A hereditary disease which is usually diagnosed in adulthood. Clinical Dermatology Review. 2021;5:200-3.
- [Google Scholar]
- A case of alkaptonuria diagnosed in the 6th decade - A clinico-dermoscopic-histopathological view. Indian Dermatol Online J. 2021;13:118-20.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Five cases of alkaptonuria among two generations of single family in Dharwad, Karnataka (India) Indian J Clini Biochemistry. 2015;30:479-84.
- [CrossRef] [Google Scholar]
- The prevalence of ochronosis in a Romani (Gypsy) population in Tamil Nadu, South India. BONE 2005 Conference Paper June :S180-S181.
- [Google Scholar]
- Mutation screening of the HGD gene identifies a novel alkaptonuria mutation with significant founder effect and high prevalence. Ann Hum Genet. 2014;78:155-64.
- [CrossRef] [PubMed] [Google Scholar]
- Founder effects of the homogentisate 1,2-dioxygenase (HGD) gene in a gypsy population and mutation spectrum in the gene among alkaptonuria patients from India. Clin Rheumatol. 2020;39:2743-9.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A.. 1977;74:5463-7.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis of alcaptonuria: Rapid analysis of homogentisic acid by HPLC. Clin Chim Acta. 1990;189:7-11.
- [CrossRef] [PubMed] [Google Scholar]
- The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611-22.
- [CrossRef] [PubMed] [Google Scholar]
- The state of RT-quantitative PCR: Firsthand observations of implementation of minimum information for the publication of quantitative real-time PCR experiments (MIQE) J Mol Microbiol Biotechnol. 2014;24:46-52.
- [CrossRef] [PubMed] [Google Scholar]
- The ultimate qPCR experiment: producing publication quality, reproducible data the first time. Trends Biotechnol. 2019;37:761-74.
- [CrossRef] [PubMed] [Google Scholar]
- Identification of reference genes in blood before and after entering the plateau for SYBR green RT-qPCR studies. Peer J. 2017;5:e3726.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Identification of reference genes for RT-qPCR data normalisation in aging studies. Sci Rep. 2019;9:13970.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- First report of a deletion encompassing an entire exon in the homogentisate 1,2-dioxygenase gene causing alkaptonuria. PLoS One. 2014;9:e106948.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Degenerative Osteoarthritis with multiple joint arthroplasties due to alkaptonuria, a rare inborn error of tyrosine metabolism. Isr Med Assoc J. 2018;20:260-1.
- [PubMed] [Google Scholar]
- The Genome Asia 100K project enables genetic discoveries across Asia. Nature. 2019;576:106-11.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Novel mutations in the homogentisate-1,2-dioxygenase gene identified in Slovak patients with alkaptonuria. J Med Genet. 2000;37:539-42.
- [CrossRef] [PubMed] [Google Scholar]
- Homogentisate 1,2-dioxygenase (HGD) gene variants, their analysis and genotype-phenotype correlations in the largest cohort of patients with AKU. Eur J Hum Genet. 2019;27:888-902.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Alkaptonuria--a review of surgical and autopsy pathology. Histopathology. 2008;53:503-12.
- [CrossRef] [PubMed] [Google Scholar]
- Amyloidosis, inflammation, and oxidative stress in the heart of an alkaptonuric patient. Mediators Inflamm. 2014;2014:258471.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Mechanisms of enhanced osteoclastogenesis in alkaptonuria. Am J Pathol. 2018;188:1059-68.
- [CrossRef] [PubMed] [Google Scholar]
- Inflammatory and oxidative stress biomarkers in alkaptonuria: Data from the Develop AKUre project. Osteoarthritis Cartilage. 2018;26:1078-86.
- [CrossRef] [PubMed] [Google Scholar]
- Alkaptonuria severity score index revisited: Analysing the AKUSSI and its subcomponent features. JIMD Rep. 2018;41:53-62.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Modifier factors of cystic fibrosis phenotypes: A focus on modifier genes. Int J Mol Sci. 2022;23:14205.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The impact of three SMN2 gene copies on clinical characteristics and effect of disease-modifying treatment in patients with spinal muscular atrophy: A systematic literature review. Front Neurol. 2024;15:1308296.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The Potential of Single Nucleotide Polymorphisms (SNPs) as biomarkers and their association with the increased risk of coronary heart disease: A systematic review. Vasc Health Risk Manag. 2023;19:289-301.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Homogentisic acid is not only eliminated by glomerular filtration and tubular secretion but also produced in the kidney in alkaptonuria. J Inherit Metab Dis. 2020;43:737-47.
- [CrossRef] [PubMed] [Google Scholar]
- Opportunities and challenges for antisense oligonucleotide therapies. J Inherit Metab Dis. 2021;44:72-87.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




