Translate this page into:
Development of ELISA based detection system for lethal toxin of Clostridium sordellii
Reprint requests: Dr S. Ponmariappan, Scientist, Biotechnology Division, Defence Research & Development Establishment Jhansi Road, Gwalior 474 002, India e-mail: ponmariappans@yahoo.com
-
Received: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
Clostridium sordellii and its toxins are associated with diseases in animals as well as human. C. sordellii produces two protein toxins (lethal toxin and haemorrhagic toxin). Lethal toxin has gained more importance due its high toxicity. The present study was carried out to develop a sandwich ELISA for detection of lethal toxin of C. sordellii.
Methods:
The catalytic domain (1.6kb) of lethal toxin of C. sordellii was PCR amplified, cloned into pQE30 UA vector and transformed into Escherichia coli SG 13009. Expression conditions were optimized and the recombinant protein was purified under native condition using Ni-NTA affinity chromatography, confirmed by SDS-PAGE and Western blot. Antibody was generated against the purified recombinant protein using Freund's complete and incomplete adjuvants (FCA and FIA) in BALB/c mice and rabbit. A sandwich ELISA was optimized for the detection of lethal toxin.
Results:
The maximum recombinant protein expression was achieved at 0.5 mM IPTG (isopropylthiogalactoside) induction 4.0 h of post-induction. The polyclonal antibody raised in mice and rabbit showed a titre up to 1:512000. The produced antibody was highly sensitive with the detection limit of 0.3 ng/ml of lethal toxin at 1:4000 dilutions of mice (capturing) and rabbit (revealing) antibody.
Interpretation & conclusions:
An ELISA based detection system was developed for the detection of lethal toxin of C. sordellii. The developed detection system was found to be specific as there was no cross-reactivity with any other clostridial toxins. It will be useful for the detection of lethal toxin of C. sordellii in clinical and environmental samples.
Keywords
Clostridium sordellii
ELISA
lethal toxin
recombinant protein
toxic shock syndrome

Clostridium sordellii is an anaerobic, Gram positive, spore forming rod which typically resides in soil and colonizes the gastrointestinal tract and genital tracts of healthy human1. It produces two antigenically different protein toxins i.e. lethal toxin (LT) and haemorrhagic toxin (HT). The lethal toxin is a single chained protein of about 270 kDa and exhibits ABCD model (A, biological activity; B, binding; C, cutting; D, delivery). It consists of four domains, receptor binding domain, catalytic domain, transmembrane domain and cysteine protease domain. The receptor-binding domain is located at the C-terminus region and responsible for binding to the target cell and its uptake by receptor mediated endocytosis. The catalytic domain is located at the N-terminus region of the toxin and causes the mono-glucosylation of low molecular weight Guanosine-5’ triphosphate (GTP) binding proteins of Rac, Cdc 42 and Ras subfamily including H-Ras, R-Ras, Ral and Rap2. The transmembrane domain is located in between these two domains and responsible for translocation of the toxin from endosome to cytoplasm. The cysteine protease domain is located in between the catalytic domain and transmembrane domain and responsible for the autocatalytic processing of lethal toxin. The catalytic domain glucosylates low molecular weight GTP binding proteins using co-substrate uridine di phosphate (UDP)-glucose and renders these inactive and inhibiting the recognition of their downstream proteins. In the case of Rac, and Cdc, modification impairs actin cytoskeleton assembly and organization leading to the massive capillary leakage which is the characteristic of C. sordellii infection3. Lung and heart are the preferential organs for action of lethal toxin of C. sordellii4.
The lethal toxin has been associated with a number of diseases including toxic shock syndrome (TSS) in post abortion and postpartum cases56, bacteremia7, toxic omphalitis in infants8, sudden death syndrome, septic arthritis9 and necrotizing fasciitis10. Recently, C. sordellii has been reported as a cause of acute constrictive pericarditis with pyopericardium and tamponade11. The list of diseases has increased with the implication of this organism in death of drug users by contaminated heroin10 and the patients receiving musculoskeletal allograft12. It causes enteritis and sudden death in sheep13141516. It also causes diarrhoea and enteric lesions in calves17. The early human clinical symptoms of C. sordellii infection are haematocrit, increased WBC, increased platelet counts, decreased serum calcium and protein levels. As infection progresses, it induces six distinct clinical features which include a marked leukocytosis known as leukemoid reaction, refractory hypotension, severe tachycardia, profound capillary leak syndrome and haemoconcentration1.
Cell culture based cytotoxicity assay is considered as the gold standard for the detection of lethal toxin. The testing time of cell culture based cytotoxicity assay requires at least 48-72 h. This disadvantage limits the usage of this diagnostic assay, in addition, maintenance of cell lines is expensive, the assay format is complex and it is difficult to obtain quantitative results. Alternate to the cell culture cytotoxicity assay, ELISA is considered as one of the most sensitive, easy and amenable method to develop a high throughput system. In the present study, an attempt was made to develop a sandwich ELSIA using polyclonal antibodies raised against the recombinant protein of lethal toxin of C. sordellii.
Material & Methods
The work was carried out in the Biotechnology Division of Defence Research and Development Establishment (DRDE), Gwalior.
Cloning and expression: The genomic DNA of C. sordellii ATCC 9714 was isolated using DNA purification kit from Promega (USA) according to the manufacturer's protocol. The forward primer LT f : 5’-AAC TTA GTT AAC AAA GCC CAA-3’ and reverse primer LT r 5’-TTA TTA TAA TAT TTT TTT AGA AAC ATA ATC-3’ specific for the catalytic domain of the lethal toxin gene were synthesized from Sigma (Bangalore) and the gene was PCR amplified. All PCR reactions18 were carried out for an initial denaturation at 94°C for 5 min followed by 30 cycles consisting of denaturation at 94°C for 1 min, annealing for 1min at 58°C, extension at 72°C for 2 min and final extension at 72°C for 10 min in a thermal cycler (Tgradient, Biometra, Germany). The amplified PCR product was checked by 1 per cent agarose gel electrophoresis. The PCR product was further purified using DNA purification kit (Qiagen, Germany) as suggested by the manufacturer's protocol. The PCR product was quantified using Nanodrop (USA). The purified PCR product was ligated with the prelinearized expression vector pQE 30 UA (Qiagen, Germany) and the chimeric vector was transformed into electro competent Escherichia coli SG 13009 using an electroporator (Gene pulser XL, Bio-Rad, USA). The positive transformants were selected on Luria Bertani (LB) agar plates (Difco, USA) containing kanamycin (30 μg/ml) and ampicillin (100 μg/ml). Plasmids were extracted from the selected transformants using QIA miniprep kit (Qiagen, Germany) following manufacture's protocol. Further, these plasmids were screened for the confirmation of the presence of inserts using LT gene specific primers mentioned above and also checked inframe using the combination of LT gene specific and pQE 30 UA vector specific primers.
Recombinant protein expression and localization: The selected transformant was inoculated into 5ml LB broth containing kanamycin (30 μg/ml) and ampicillin (100 μg/ml) and grown overnight at 37°C with constant shaking. One per cent of the overnight grown culture was further inoculated in 30 ml LB broth containing respective antibiotics and grown at 37°C. Growth was monitored by measuring the absorbance OD600. Approximately at OD600 0.6 -0.8 the recombinant protein expression was induced with different concentrations (0.25, 0.5, 0.75, 1 mM) of isopropylthiogalactoside (IPTG) and the growth was resumed for 4.0 h under the same conditions. Prior to IPTG induction, 2 ml aliquots were taken out aseptically to be used as uninduced control. After induction, 2ml samples were withdrawn at every one hour interval and cells were harvested by centrifugation at 8000 × g for 10 min at 4°C. Each sample was analyzed by 12 per cent SDS -PAGE using molecular weight marker (Fermentas, Canada). Localization of the recombinant protein was done by solubilizing the induced cell pellet in phosphate buffer (50mM NaH2 PO4, 300 mM NaCl, 10m Mimidazole) at the ratio of 1:5 (w/v). Lysozyme (1mg/ml) was added to the cell lysate and it was kept in ice for 30 min and then frozen at -20 °C. After thawing it was sonicated by 9 sec pulses with 9 sec pause for 10 min. The cell lysate was centrifuged at 10000xg for 40 min at 4°C and supernatant and pellet were checked for the presence of the recombinant protein on 12 per cent SDS-PAGE19.
Purification of recombinant lethal toxin (rLT) protein: Supernatant was used for the purification of recombinant lethal toxin protein using affinity chromatography. One ml of Ni-NTA slurry (Qiagen, Germany) was packed into the column and it was equilibrated using the equilibration buffer (50 mM NaH2PO4, 300 mM NaCl, and 10 mM imidazole) and 5 ml of supernatant (supernatant + 20mM β-ME, 2% Triton X-100) was loaded on to it. Column was left for 30 min at room temperature with lysate and in between Ni-NTA slurry was mixed gently to allow binding. The column was washed with 10-12 volumes of wash buffer (50 mM NaH2PO4, 300 mM NaCl, 60 mM imidazole and 2% Triton) and then the protein was eluted with elution buffer (250 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole). Different elutes were collected and checked on 12% SDS-PAGE along with different washes and flow through. The collected elutes were pooled together and dialyzed against the phosphate buffer saline (PBS). Finally, the dialysed protein was concentrated using an Amicon ultra cell with 30kDa cut off membrane (Millipore, USA). The protein concentration was determined using BCA protein assay kit (Sigma, USA).
Western blot analysis: The rLT protein is tagged with His6 fusion protein and to confirm the presence of purified protein, commercially available anti-His monoclonal antibody (Qiagen, Germany) was used. Purified recombinant protein was run on 12 per cent denaturing gel (SDS-PAGE), along with prestained molecular weight marker (MBI Fermentas, Canada) on adjacent lanes and transferred electrophoretically to the nitrocellulose membrane (Millipore, USA). The membrane was incubated in 3 per cent bovine serum albumin (BSA, fraction V) in PBS at 4°C for overnight. The nitrocellulose membrane was washed three times with PBS containing 0.05 per cent Tween-20 (PBS-T) and once with PBS and then incubated at 1:1000 dilution of mouse anti-His-monoclonal antibody for 1.0 h with gentle shaking. The membrane was again washed three times with PBS-T and once with PBS and then the membrane was incubated in 1:2000 dilution of rabbit anti-mouse IgG horseradish peroxidase (HRP) (Dako, Denmark) as secondary antibody with gentle shaking at room temperature for 1.0 h. The membrane was washed and the protein bands were visualized by incubating the membrane in 3,3’-diaminobenzidine in PBS containing 8.8mM H2O220.
Antibody generation: The study protocol was approved by the Institutional Ethics Committee of DRDE. BALB/c mice (approximately 16-22 g), Australian white rabbit (approximately 1-2-1.8 kg) were selected for immunization. The rLT purified under native conditions was used for the production of polyclonal antibody in both mice and rabbit. A group of eight mice was immunized intraperitonially with 10μg and one rabbit was immunized subcutaneously with 100 μg of purified rLT with Freund's complete adjuvant (FCA) (Sigma, Aldrich, USA). The priming was followed by booster immunizations on days 7, 14, 21 days with increasing concentrations of protein with Freund's incomplete adjuvant (FIA) (Sigma, Aldrich, USA). Total volume used per mouse at each immunization was 100 μl and rabbit 500 μl. Blood was collected from retro-orbital plexus of mice with heparinised capillaries and from the heart of the rabbit before the first dose and four days after the last dose. The collected blood was incubated at 37°C for 30 min and subsequently incubated at 4°C for 1 h. The serum was collected after the centrifugation of the blood samples at 6000 × g for 10 min to get rid of residual blood cells. All serum samples were stored at -80°C for further studies.
Indirect enzyme-linked immunosorbent assay (ELISA): The collected serum was tested for the recognition of rLT by ELISA. Microtitre plates (Nunc-Immuno plate with Maxisorp surface, Nunc, Roskilde, Denmark) were coated with 500ng/well of rLT protein in coating buffer (0.05M carbonate-bicarbonate buffer pH 9.6) and incubated overnight at 4°C. The plates were blocked with 300μl of 3 per cent BSA in PBS for 2.0 h at 37°C. Plates were washed three times with PBS-T and once with PBS. Mouse anti-rLT and rabbit anti-rLT test serum samples were serially diluted to two-fold in 1 per cent BSA in PBS starting from 1:1000 to 1: 20,48,000 and incubated in duplicate wells (100 μl/well) at 37°C for 1.0 h. The plate was washed as above. The rabbit anti-mouse IgG-HRP (Dako, Denmark) and goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology, USA) diluted 1:2000 in 1 per cent BSA in PBS were added to the wells (100 μl/well) and incubated at 37°C for 1.0 h. After washing, the plates were developed by adding 2, 2’-azino-bis(3-ethylbenzo-thiazoline-6-sulphonic acid) diammonium salt solution containing H2O2 and it was incubated at 37°C for 25 min (100 μl/well). The absorbance was measured at 410 nm (A 410) using an ELISA plate reader (Biotek, USA). Mean A 410 values and standard errors of the mean (SEM) for each group were calculated. Dilutions of the serum samples at which the absorbance value was two times the cut-off value (A 410 using the preimuune serum) were considered to be the ELISA endpoint. The cut-off value for assay was calculated as the mean specific optical density plus three times the standard deviation (SD) for pre-immune serum assayed at a dilution of 1:1000. The cross-reactivity of the raised antibody with other clostridial species was checked by coating the microtitre plates with 2μg of the trichloroacetic acid (TCA) precipitated culture supernatant of C. sporogenes ATCC-11437, C. perfringens ATCC-13124 (from ATCC, USA) C. tetani 49205 (from CRI, Kasauli), C. difficile, Staphylococcus aureus and Shigella dysenteriae (from culture collection Biotechnology Division, DRDE, Gwalior). For S. aureus and S. dysenteriae cell lysates were used.
Sandwich enzyme-linked immunosorbent assay (ELISA): The optimum dilutions of mice and rabbit anti-rLT serum were determined through checkerboard titration and 1:4000 dilutions were found suitable for sandwich ELISA. Further, the raised mice anti-rLT antibody was diluted to 1:4000 in coating buffer and used to coat the polystyrene wells (100μl/well) in duplicates and incubated overnight at 4°C. After decanting the wells, the remaining sites of adsorption were blocked by addition of 3 per cent BSA in PBS, 300 μl/well and incubated for 2 h at 37°C. Then the wells were washed three times with PBS-T and once with PBS. Purified rLT (100 μl/well) serially diluted (10 μg to 0.0375 ng/ml) in PBS containing 1 per cent BSA was added to the blocked antibody coated wells and incubated for 1 h at 37°C. The wells were washed as mentioned above. Again the wells were incubated with raised rabbit anti-rLT antibody (100 μl/well) at the dilution of 1:4000 in 1 per cent BSA in PBS and incubated at 37°C for 1 h. The wells were washed as above and further incubated at 37°C for 1 h with the HRP-conjugated goat anti-rabbit IgG at a dilution of 1:2000. The wells were washed three times with PBS-T and once with PBS. The cut-off value for assay was calculated as the mean specific optical density plus three times the standard deviation (SD) of the well having no antigen. Concentration of the recombinant lethal toxin at which the absorbance value was two times the cut-off value (A 410 using the well having no antigen) was considered to be the ELISA endpoint.
Results
Cloning, expression and purification of recombinant protein: The amplified PCR product (1.6 kb) was highly specific to the catalytic domain of the lethal toxin gene of C. sordellii, was confirmed by double pass sequencing followed by multiple sequence alignment with standard lethal toxin gene sequence available in NCBI database. The purified PCR product was cloned in pQE30 UA expression vector which is having a Histidine tag in the N-terminal end and aids in the purification and detection of recombinant lethal toxin protein. After ligation, the recombinant vector was transformed into the E. coli SG 13009. Plasmid was isolated from the selected transformant and checked for the presence of insert using LT gene specific forward and reverse primers. The recombinant protein expression conditions were optimized. The maximum yield of recombinant protein was obtained at OD600 0.6 with 0.5 mM IPTG induction with minimum incubation of 4.0 h after induction. Thereafter, the cells were harvested and subjected to localization studies. The results revealed that the majority of the recombinant lethal toxin protein was present in the supernatant and was confirmed by Western blot analysis with anti His antibody. The SDS-PAGE of the localized protein and its Western blot are shown in Fig. 1a and b. The Western blot analysis clearly showed a single band size of approximately 63 kDa which is the expected size of the lethal toxin fragment. Further efforts were made to purify the protein under native conditions. Various combinations of binding and wash buffer were used in a Ni-NTA affinity column and the protein was purified near to homogeneity under native conditions. The SDS-PAGE of the purified recombinant protein is shown in Fig. 2a and the Western blot is shown in Fig. 2b.
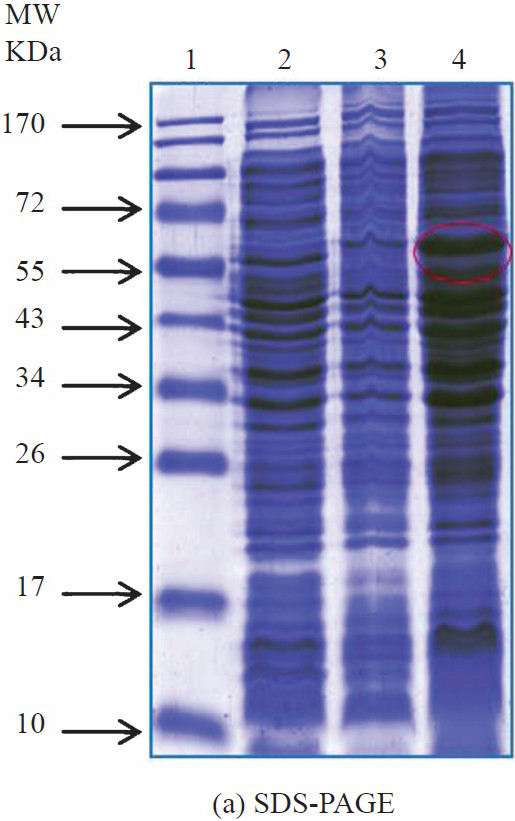
- SDS-PAGE profile for the localization of recombinant lethal toxin protein. Lane 1- Protein molecular weight marker(kDa); Lane 2: E. coli SG 13009 (uninduced); Lane 3: Induced cell pellet after sonication; Lane 4: supernatant after sonication.

- Western blot for the localization of recombinant lethal protein. Lane 1- Protein molecular weight marker (kDa); Lane 2: E. coli SG 13009 (uninduced); Lane 3: Induced cell pellet after sonication; Lane 4: supernatant after sonication.

- Affinity purified recombinant lethal toxin protein under native conditions. Lane 1: Protein molecular weight marker; Lane 2-10: elutes.

- Western blot of purified recombinant lethal toxin protein using anti-His antibody. Lane 1: Protein molecular weight marker; Lane 2: purified recombinant lethal toxin protein.
Antibody generation: The hyperimmune serum was raised in mice and rabbit against the recombinant lethal toxin protein and the serum titre was estimated using plate ELISA by coating 500ng/well of purified recombinant lethal toxin protein. The titre upto 1:5 12000 was found in both mice and rabbit with the cut-off value of >0.085 and 0.087, respectively. No cross-reactivity was observed with TCA precipitated culture supernatant of C. difficile, C. sporogenes, C. perfringens and C. tetani. Similarly, no cross-reactivity was observed with cell lysate of S. aureus and S. dysenteriae.
Evaluation of detection limit: The recombinant protein was assayed in the concentration ranges from 10 μg to 0.0375 ng/ml to determine the limit of detection in the ELISA format. Mice anti-rLT specific antibody was used to capture the antigen at 1:4000 dilution and rabbit anti-rLT specific antibody were used to reveal the antigen at 1:4000 dilutions. Results showed that the rLT could be detected up to a concentration of approximately 0.3 ng/ml with the cut-off value of >0.129 (Fig. 3).

- Minimum detection limit of lethal toxin of C. sordellii by sandwich ELISA. OD values represented are mean±SD of triplicate determinations.
Discussion
C. sordellii is an important human pathogen which causes a number of diseases. The most fatal among these is fulminant toxic shock syndrome in post partum and post-abortion cases. In Canada, the number of medically induced abortions (MIA) associated with mifepristone and misoprostal cases has been shown to be increasing21. Apart from that, ten cases of soft tissue infections among injection drug users (IDU) were reported at different time period which were mainly caused by C. sordellii10.
Sensitive and rapid detection of lethal toxin is essential to save the life of the patients. Currently, the most reliable method for detection of lethal toxin is cell culture based cytotoxicity assay22, while this assay is sensitive but it is slow, expensive and requires animal cell culture facilities and skilled manpower to perform the tests. As such there is no commercial detection system available for the specific detection of lethal toxin. In the present study, we developed an ELISA based detection system alternate to the in vitro system for the detection of lethal toxin in clinical and environmental samples.
Only a few studies have been carried out on lethal toxin of C. sordellii, the native lethal toxin (LT) was purified from the culture supernatant of C. sordellii IP82 by DEAE-Trisacryl, Ultrogel AcA3-4 gel filtration and hydroxyapatite column chromatography. The molecular weight of purified LT was estimated to be 240 to 250kDa and the isoelectric point (pI) was 4.55. The study concluded that antibodies raised against C. sordellii LT neutralized C. difficile toxin B23. Cloning of the gene enabled us to have the sufficient amount of the protein without handing the pathogenic culture and it facilitated its purification as a tagged protein. In the present study, a fragment of lethal toxin gene was cloned in pQUE30 UA and expressed in E. coli SG 13009 to develop an ELISA based detection system. Similarly, Dreger et al24 have cloned the gene encoding for the catalytic domain of lethal toxin of C. sordellii in pGex expression system with GST-fusion protein to prove that the cytotoxic effects of TcsL depend on the glucosylation of critical substrate proteins rather than on the glucosyltransferase activity. Mesmin et al18 cloned the catalytic domain into pET-28 and expressed as His tagged protein to show the glucosyltransferase domain of lethal toxin of C. sordellii.
In the present study, we produced a high titre (1:512000) antibody which did not show any cross-reactivity with C. difficile and other closely related clostridia toxins. Similar kind of assay was developed from the inactivated whole lethal toxin of C. sordellii and the antibody showed cross reactivity with C. difficile toxin B but in the meantime it can also neutralize the cytotoxicity of C. difficile23. The raised capture antibody against the catalytic domain of lethal toxin of C. sordellii was able to detect the lethal toxin up to concentration of approximately 0.3 ng/ml. The developed ELISA system is specific, rapid and can be used for the testing of a large number of samples. The more sensitive and rapid molecular techniques have been used for the detection of C. sordellii and lethal toxin gene from clinical specimens. Valour et al25 isolated C. sordellii from brain abscess and confirmed by 16S rRNA gene sequencing. Highet et al26 made an attempt for the direct PCR based detection of C. sordellii lethal toxin gene from the intestinal flora of infants who died from the sudden infant death syndrome but failed to detect by PCR. These methods are ultrasensitive in pure culture based studies, but when it comes to the clinical and environmental samples, these techniques fail to detect toxin due to the presence of PCR inhibitors. It requires expertise to clean up the samples to get true positive results.
In conclusion, an antibody based immuno detection system was developed for the detection of lethal toxin of C. sordellii. The generated high titre antibody will be highly useful for the detection of lethal toxin of C. sordellii from clinical and environmental samples.
Acknowledgment
Authors thank the Director, DRDE, Gwalior, for providing necessary facilities and support required for this study. Authors are also thankful to Dr Pravin Kumar, Animal Facility of DRDE, Gwalior, for providing animals.
References
- Clostridium sordellii infection: epidemiology, clinical findings, and current perspectives on diagnosis and treatment. Clin Infect Dis. 2006;43:1436-46.
- [Google Scholar]
- Clostridial Rho-inhibiting protein toxins. Curr Top Microbiol Immunol. 2005;291:113-45.
- [Google Scholar]
- Difference in protein substrate specificity between hemorrhagic toxin and lethal toxin from Clostridium sordellii. Biochem Biophys Res Commun. 1996;229:370-4.
- [Google Scholar]
- Clostridium sordellii lethal toxin kills mice by inducing a major increase in lung vascular permeability. Am J Pathol. 2007;170:1003-17.
- [Google Scholar]
- Toxic shock syndrome due to Clostridium sordellii: a dramatic postpartum and postabortion disease. Clin Infect Dis. 2002;35:1441-3.
- [Google Scholar]
- Postpartum Clostridium sordellii infection associated with fatal toxic shock syndrome. Acta Obstet Gynecol Scand. 2000;79:1134-5.
- [Google Scholar]
- Clostridium sordellii bacteremia: case report and review. Clin Infect Dis. 1992;15:950-4.
- [Google Scholar]
- Neonatal Clostridium sordellii toxic omphalitis. Pediatr Infect Dis J. 1993;12:253-7.
- [Google Scholar]
- Polymicrobial septic arthritis due to Clostridium species: case report and review. Clin Infect Dis. 2000;30:590-4.
- [Google Scholar]
- Outbreak of necrotizing fasciitis due to Clostridium sordellii among black-tar heroin users. Clin Infect Dis. 2004;38:e87-91.
- [Google Scholar]
- Clostridium sordellii as a cause of constrictive pericarditis with pyopericardium and tamponade. J Clin Microbiol. 2011;49:3700-2.
- [Google Scholar]
- Clostridium infections associated with musculoskeletal-tissue allografts. N Engl J Med. 2004;350:2564-71.
- [Google Scholar]
- Sudden death in periparturient sheep associated with Clostridium sordellii. Vet Rec. 2003;153:340.
- [Google Scholar]
- Sudden death in sheep associated with Clostridium sordellii. Vet Rec. 1998;142:417-21.
- [Google Scholar]
- Effect of booster vaccination with a multivalent clostridial bacterin-toxoid on sudden death syndrome mortality rate among feedlot cattle. J Am Vet Med Assoc. 1997;211:749-53.
- [Google Scholar]
- Production od diarrhoea and enteric lesions in calves by the oral inoculation of pure cultures of Clostridium sordellii. Vet Rec. 1983;112:141-6.
- [Google Scholar]
- A phosphatidylserine-binding site in the cytosolic fragment of Clostridium sordellii lethal toxin facilitates glucosylation of membrane-bound Rac and is required for cytotoxicity. J Biol Chem. 2004;279:49876-82.
- [Google Scholar]
- Cleavage of structural proteins druing the assembly of the head of bacteriophage T4. Nature. 1970;227:680-5.
- [Google Scholar]
- Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350-4.
- [Google Scholar]
- Clostridium sordellii toxic shock syndrome after medical abortion with mifepristone and intravaginal misoprotol - United States and Canada, 2001-2005. MMWR Morb Mortal Wkly Rep. 2005;54:724.
- [Google Scholar]
- Comparative study of immunological properties and cytotoxic effects of Clostridium difficile toxin B and Clostridium sordellii toxin L. Toxicon. 1992;30:129-40.
- [Google Scholar]
- Purification and characterization of Clostridium sordellii lethal toxin and cross-reactivity with Clostridium difficile cytotoxin. Infect Immun. 1987;55:35-43.
- [Google Scholar]
- Killing of rat basophilic leukemia cells by lethal toxin from Clostridium sordellii: critical role of phosphatidylinositide 3’-OH kinase/Akt signaling. Biochemistry. 2009;48:1785-92.
- [Google Scholar]
- Clostridium sordellii brain abscess diagnosed by 16S rRNA gene sequencing. J Clin Microbiol. 2010;48:3443-4.
- [Google Scholar]
- Clostridium sordellii lethal toxin gene is not detectable by PCR in the intestinal flora of infants who died from sudden infant death syndrome or other causes. J Med Microbiol. 2010;59:251-3.
- [Google Scholar]




