Translate this page into:
Cytogenomics of hexavalent chromium (Cr6+) exposed cells: A comprehensive review
Reprint requests: Dr Sushil Kumar, Environmental Carcinogenesis Division, CSIR-Indian Institute of Toxicology Research Mahatma Ganghi Marg, Post Box 80, Lucknow 226 026, India e-mail: sushilkumar@iitr.res.in
-
Received: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The altered cellular gene expression profile is being hypothesized as the possible molecular basis navigating the onset or progress of various morbidities. This hypothesis has been evaluated here in respect of Cr6+ induced toxicity. Several studies using gene microarray show selective and strategic dysregulations of cellular genes and pathways induced by Cr6+. Relevant literature has been reviewed to unravel these changes in different test systems after exposure to Cr6+ and also to elucidate association if any, of the altered cytogenomics with Cr6+ induced toxicity or carcinogenicity. The aim was to verify the hypothesis for critical role of altered cytogenomics in onset of Cr6+ induced biological / clinical effects by identifying genes modulated commonly by the toxicant irrespective of test system or test concentrations / doses, and by scrutinizing their importance in regulation of the flow of mechanistically linked events crucial for resultant morbidities. Their probability as biomarkers to monitor the toxicant induced biological changes is speculative. The modulated genes have been found to cluster under the pathways that manage onset of oxidative stress, DNA damage, apoptosis, cell-cycle regulation, cytoskeleton, morphological changes, energy metabolism, biosynthesis, oncogenes, bioenergetics, and immune system critical for toxicity. In these studies, the identity of genes has been found to differ remarkably; albeit the trend of pathways’ dysregulation has been found to remain similar. We conclude that the intensity of dysregulation of genes or pathways involved in mechanistic events forms a sub-threshold or threshold level depending upon the dose and type (including speciation) of the toxicant, duration of exposure, type of target cells, and niche microenvironment of cells, and the intensity of sub-threshold or threshold level of the altered cytogenomics paves way in toxicant exposed cells eventually either to opt for reversal to differentiation and growth, or to result in toxicity like dedifferentiation and apoptosis, respectively.
Keywords
Apoptosis
chromate carcinogenesis
chromium
epigenetics
genomics
microarray

Introduction
Hexavalent chromium (Cr6+) is a toxic metal known for its carcinogenic effect in humans. The lung cancer risk is prevalent in pigment chromate handlers, ferrochromium production workers, stainless steel welders, and chromeplaters1. Besides occupational cancer, risk of other kinds of adverse health effects are also reported in humans after short term/prolonged exposure through inhalation, ingestion, or topical contact2345.
Chromate compounds are cytotoxic, genotoxic, and carcinogenic in nature678910111213141516171819. Mechanism of action is proposed to involve reactive oxygen species (ROS) generation, oxidative stress, and DNA damage; a variety of other changes like increased formation of DNA adducts and DNA-protein cross-links, DNA strand breaks, chromosomal aberrations and instability20212223242526, disruption of mitotic cell division, chromosomal aberration, premature cell division262728293031323334, S or G2/M cell cycle phase arrest353637383940414243, and carcinogenesis44 also occur in humans or experimental test systems. However, the molecular basis of these changes culminating into biological effects, tissue lesions, or cancers have not been examined. Cr6+ induced alterations in cellular gene expression forming the basis of these changes remain an unexplored probability. Several cytogenomic studies using gene microarray approach demonstrated a selective and strategic dysregulations of cellular genes and pathways by Cr6+. The cytogenomics studies gave toxicologists a comprehensive view of genomic changes occurring in the pathogenesis of Cr6+ toxicity.
The aim of this review was to derive a hypothesis on the critical role of altered cytogenomics and its intensity culminating into the elicited biological/clinical effects, and to verify this hypothesis by (i) identifying genes and pathways modulated commonly by Cr6+ irrespective of test system or exposure conditions, and (ii) by scrutinizing their importance in regulation of the flow of mechanistically linked intracellular events that may be crucial for Cr6+ induced toxicity or carcinogenicity.
Oncogene activation, DNA adduct formation, select gene expression, and epigenetic changes
Earlier non-microarray based studies investigated the role of mutations in oncogene like ras, p53, Bcl-2, cyclin-D1 or their altered expression in Cr6+ carcinogenesis; these studies were conducted in experimental test systems or cancer tissues of Cr6+ exposed workers. Activated ras oncogene was seen in Cr6+ lung cancer, however, considered a rare event and not involved in Cr6+ carcinogenesis45. Changes in Bcl-2 and p53 expression level were noted although these were found to be unspecific to Cr6+ carcinogenesis; the study was inconclusive as the levels were found to be similar in cancer tissue from ex-chromate workers as well as the non-exposed subjects and workers with pneumoconiosis45. Further investigations revealed mutant p53 gene in lung cancer of chromate exposed workers46 illustrating p53 mutation following Cr6+ exposure; the elevated serum levels of pantropic p53 (pan-p53) proteins in Cr6+ workers47; and induction of p53 level up to 6-fold in Cr6+ exposed human lung fibroblasts48. The key role of p53 gene in chromate toxicity or carcinogenesis was demonstrated using p53 deficient transgenic mice4950; intervention studies showed that the loss of crucial gene p53 increased the genomic DNA fragmentation49.
Recently, the effect of short term high dose (0.05 and 0.25 μM) Cr6+ exposure on benzo alpha pyrene (B(a)P) (DNA damage) directed gene alteration in mouse hepatoma cells was investigated51 RT-PCR based analysis showed upregulation in genes related to apoptosis (Aifm, Bid, Bak, Bcl2, Fas, Apaf1, Tnf, Bax), cell cycle control (Rad17, Mdc1), tumour suppression (p15, p16, p18, p19, p21, p27), DNA damage (Brca1, Brca2, ATM, Gadd45, Mgmt) and down-regulation in genes related to drug metabolism (Cyp1b1, Cyp1z2, Gsta1, Nqo1, Cyp1a1, Aldh3). In an in vivo study using mice exposed to (0, 50, 500 and 5000 ppb) Cr6+ in drinking water for two months and co-exposed to B(a)P for 24 h, downregulation of all the genes except Cyp1b1 gene in Cr6+ exposed mouse liver was seen51. In an earlier study, the co-exposure of Cr6+ and B(a)P was found to increase the carcinogen-DNA adduct formation in mouse hepatoma cells52. These observations indicated that Cr6+ exposure facilitated the carcinogen - DNA adducts formation causing DNA damage.
With respect to epigenetic changes, Cr6+ induced methylation of p16 promoter and repression of DNA-mismatch-repair or tumour suppressor genes mut L homologue 1(MLH1) and MLH2 has been reported5354 besides the genetic instability in chromate lung cancer. Sun et al55 reported an increase in protein as well as mRNA level of G9a, a histone methyl-transferase that methylated H3K9 (histone H3 lysine 9) and accounted for global elevation of its dimethylated type and silencing of tumour suppressor gene MLH1 transcription. Others showed that Cr6+ inhibited the transcription co-activators5657. Klein et al58 showed methylation of genes and modulation of gene cyclin-D1 by Cr6+ in transgenic cells; study revealed the responsiveness of cell cycle regulation to the toxic metal. A crucial role of cyclin D1 in Cr6+ toxicity was noticed in a study on ex-chromate workers affected with lung cancer wherein cyclin-D1 expression was found to be more as compared to non-exposed subjects harbouring other disease like pneumoconiosis45. The altered expression of ATM (ataxia telangiectasia mutated) gene59, aneuploidy and dysregulation in spindle assembly checkpoint bypass60 were reported in Cr6+ exposed cells; these changes normally support apoptosis, cell cycle regulation, as these are requisites of cells responding to DNA damage and to genomic instability.
Studies demonstrated alterations in cellular pathways after Cr6+ exposure. In cell signalling (MAPK) pathway, activation of (Extra cellular signal regulated kinase) ERK, (C-Jun-N-terminal kinase) JNK, (mitogen activated protein kinase) p38 (regulators of cell growth, proliferation, apoptosis, and differentiation.) was observed; the activation of change depended on toxicant's concentrations, resultant ROS generation or oxidative stress616263646566. Their activation was also reported in Cr6+-exposed mouse embryonic stem cells67; lower level of toxicant activated JNK (c-Jun-N-terminal kinase) via LCK (leukocyte C-terminal Src kinase, a member of the Src family of protein tyrosine kinases) or the Fyn-Cas-Crk (FAK/Src-Yes-Fyn/p130 CAS/CRK) signalling cascade; LCK could activate STAT3 (signal transducer and activator of transcription) and (interleukin-6) IL-6 which contributed to inflammation and cancer68. Others studies investigating ROS dependent changes found that Cr6+ exposure activated nuclear factor kappaβ (Nfkβ) and p38 (mitogen activated protein kinase 14) pathway; Nfkβ, important for apoptosis, was also considered an indicator of Cr6+ induced cytotoxicity6970. Using cultured cells, investigators also showed activation of activator protein-1 (AP-1) but HOGG1 (8-oxoguanine DNA glycosylase) gene was found to be uninfluenced. It is inferred that Nfkβ does not participate in tumourigenesis; it is rather associated with a decrease in cell proliferation and induction of apoptosis71. Overexpression of inflammation specific COX-2 via Nfkβ / c-Jun / AP-1 dependent pathway was observed in normal human bronchial epithelial cells and mouse embryonic fibroblasts after Cr6+ exposure72. The signalling molecule (VEGF) vascular endothelial growth factor was found to be overexpressed by Cr6+. VEGF, involved in angiogenesis, is usually overexpressed in lung cancer, and used as prognostic marker7374757677; one study78 on the contrary showed the suppression of VEGF expression by Cr6+. In signalling pathway, other types of genes that are activated in response to Cr6+ are Fyn and LCK and the initiation of an interferon signalling mechanism6979. Activation of AKT (α serine-threonine protein kinase) was also noticed by Cr6+ in human lung fibroblast transformation. AKT is known to override G1/S checkpoint bypass, prevent Cr6+ induced decrease in localization of retinoblastoma protein and p27 (cycline dependent kinase inhibitor 1B) the key factors of G1/S checkpoint, and contribute to toxicant induced genomic instability80. Levels of ApoJ / CLU (a senescence biomarker apolipoprotien J and an oxidative stress responsive gene protein clusterin) in serum were noted to be high in shipyard welders during the oxidative stress and were found to be lower after worksite intervention81.
The sporadic studies on oncogene activation, gene expression with or without DNA adduct formation, and epigenetic changes provided only a limited knowledge on the role of mutagenic events, oncogenes and tumour suppressor genes, and the concurrent changes in expression of assorted genes in Cr6+ carcinogenesis. To elucidate comprehensive information on the change in cytogenomics after Cr6+ exposure, the investigators used gene microarray based approach. These efforts were made in conjunction with the hypothesis that the mechanism of Cr6+ carcinogenesis was not limited only to Fenton-reactions, or the resultant genotoxic effects, or the oncogenes / tumour suppressor genes, but also involved critical alterations in global gene expressions. Both in vivo and in vitro studies elucidated epigenetic and gene expression changes that logically seemed crucial for shaping sub-clinical effects of Cr6+ like inflammation, apoptosis, and cell transformation. These exploratory studies used the limited gene-microarray or whole genome microarray, different test systems / test concentrations / test compounds / exposure durations. These investigations yielded an explosion of information for its utility to understand the key gene expression changes that contribute to toxicity after Cr6+ exposure or form the molecular basis of its toxic effects.
This review reveals the identity of dysregulated genes and pathways that could form the molecular basis of Cr6+ toxicity, and be useful in elucidating the mechanism of action of this toxicant.
Genomic studies
The microarray-based in vivo / in vitro studies8283848586878889 are summarized in this section. In these investigations, sodium or potassium dichromate, or sodium chromate were used in test systems of human cell i.e. peripheral blood mononuclear cells (PBMC), A549, BEAS-2B, dermal fibroblasts or in rat. The test concentrations ranged from 9-300μM in case of cells; a dose of 0.25 mg/kg body wt was used for rat. The size of microarrays included 216, 1200, 2400, 12000, 22000, 28000 or 44,000 probes. These studies examined genes and relevant pathways. Irrespective of the duration of toxicant exposure, stress and apoptosis were found to be the most influenced pathways; energy metabolism, DNA repair / metabolism, biosynthesis, and oncogene were moderately influenced; immunoregulation and the cell and focal adhesion / gap and tight junction / extracellular matrix / cytoskeleton were mildly influenced pathways.
In vitro studies
A549 cells, 300 μM potassium dichromate, 2 h exposure: In the first study on Cr6+ induced cellular gene expression modulation, Ye and Shi82 examined genomics in human lung type II epithelial A549 cell using microarray of 2400 genes and potassium dichromate as a source of Cr6+. They investigated the molecular basis of Cr6+ provoked ROS generation and the resultant oxidative stress, and found a significant dysregulation of 220 genes that were part of the pathways of oxidative stress, Ca2+ mobilization, energy metabolism, protein synthesis, cell cycle regulation, apoptosis, and carcinogenesis.
In stress response pathway (Table I), an upregulated transcription was seen in Cu / Zn superoxide dismutase (SOD), glutathione peroxidase, MT-IIA, MTF-1 (metal-regulatory transcription factor), p53, heat shock proteins (HSP60, HSP70, HSP75), and activating transcription factor-3 (ATF-3). The oxidative stress responsive proteins protected the correct conformation of newly formed proteins; their main function was to protect cells from ROS / oxidative stress and preserve the vitality of cells.
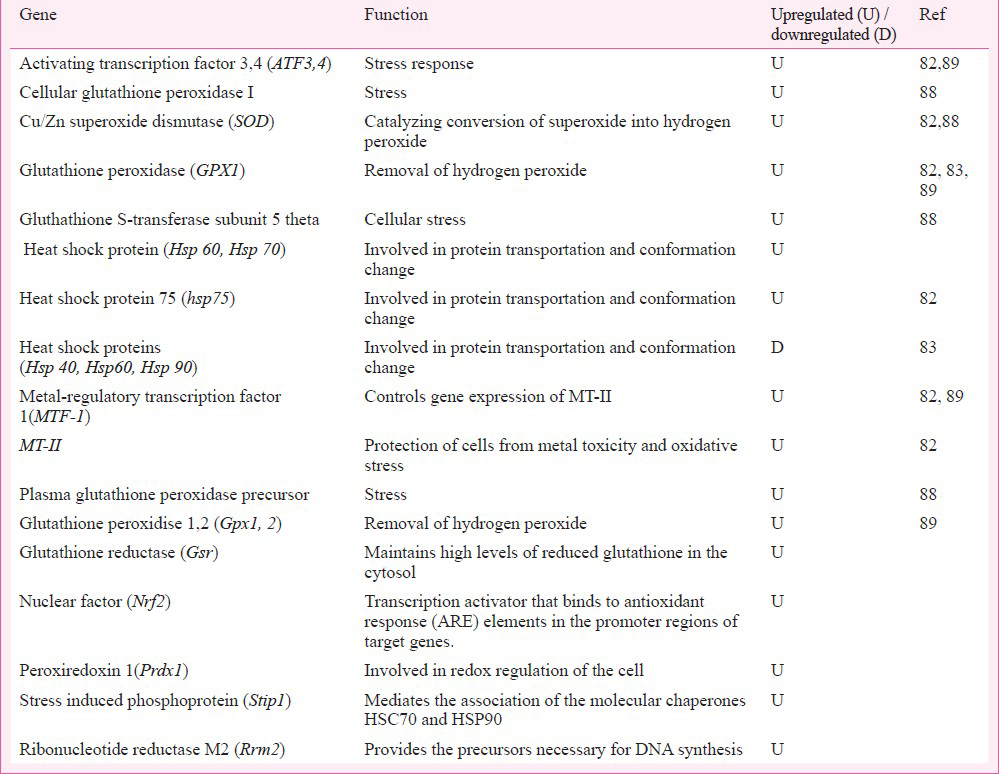
In apoptosis pathway Ye and Shi82 reported upregulation of only the hSIAH1 (Siah E3 ubiquitin protein ligase 1) gene (Table II). Functionally, this apoptotic gene facilitated to label the protein for proteasomal degradation and the programmed cell death through induction of p53 signalling during Cr6+ induced stress.

In cell cycle regulatory pathway (Table III), the upregulated genes transcribed protein products important for cell survival, cell polarization, cell growth and differentiation, G1 cell cycle arrest, and tumour suppressor function. By underexpressing the respective proteins, the downregulated genes dysregulated cell cycle control via slowdown of cyclin dependent kinase activation causing cell cycle arrest, and challenging epithelial cell integrity for apoptosis.

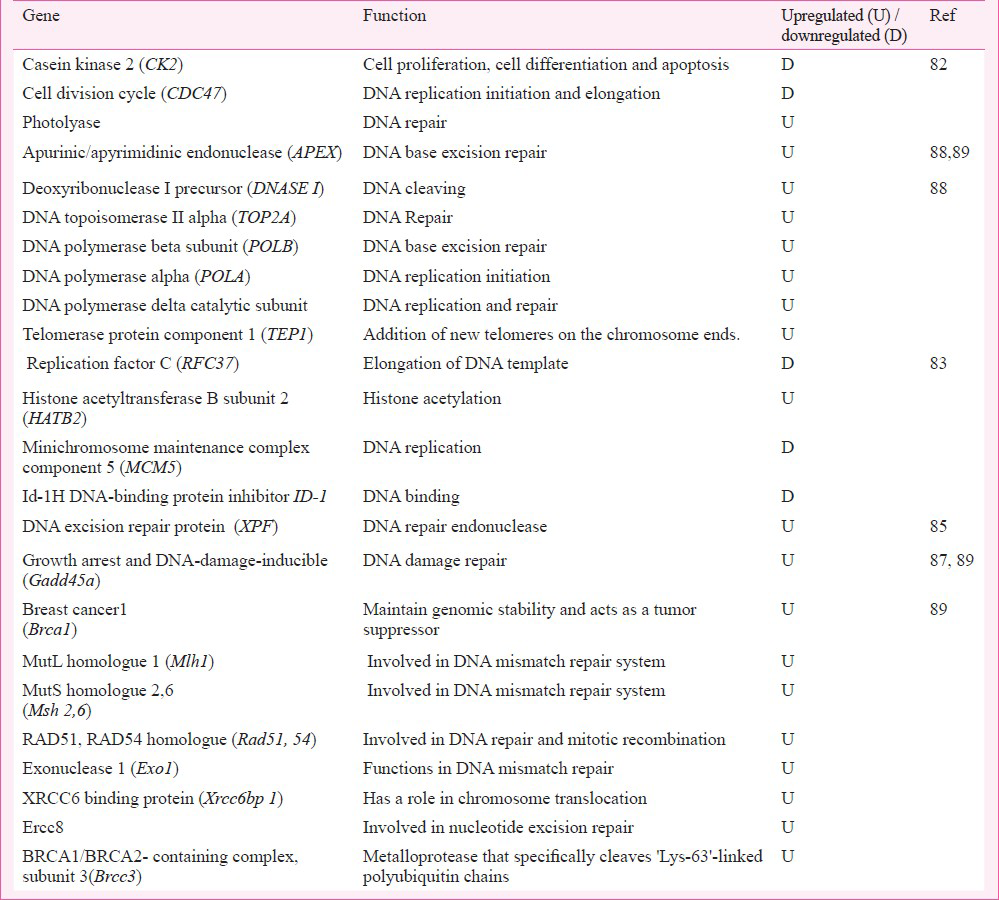
In DNA repair and metabolism pathway (Table IV), only three genes were found to be dysregulated. The upregulated gene encoded photolyase that was involved in DNA repair. The downregulated CK2 (casein kinase 2) and cell division cycle (CDC)47 decreased the function of serine / threonine kinase and also required for DNA replication. The binding of CDK4 (cell cycle dependent kinase), CDK5 with CK2/CDC45 encoded a protein to regulate the signalling mechanisms in cell proliferation and growth.
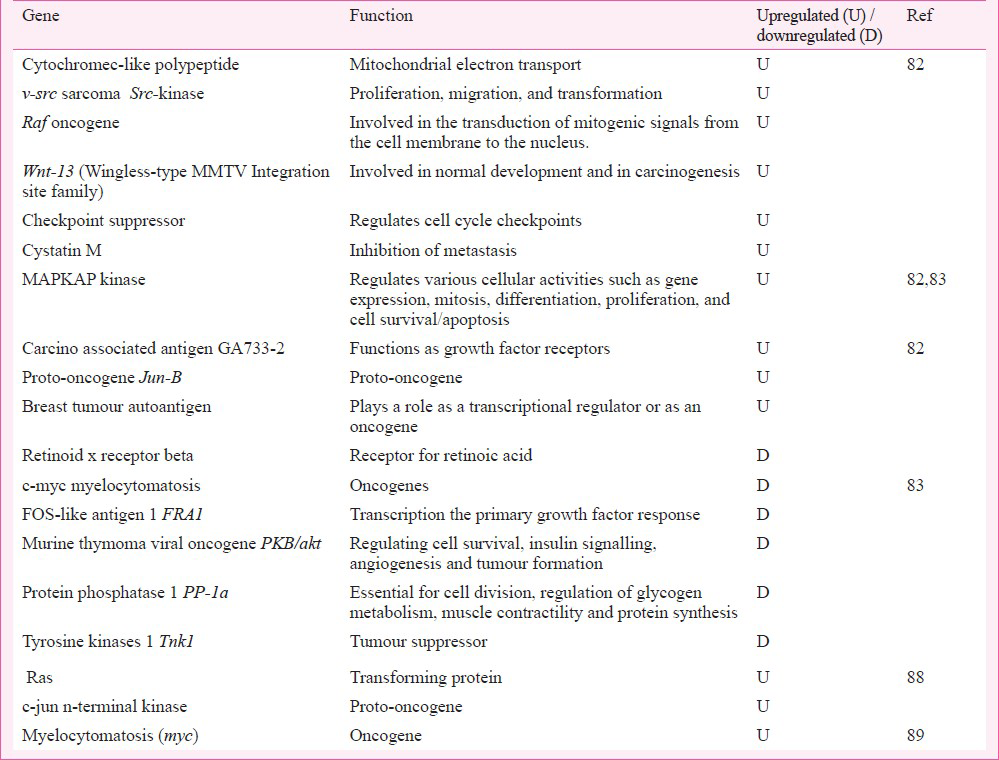
Among oncogenes (Table V), the upregulation of intracellular kinase, G-protein, Src, and MAPK showed their involvement in cell proliferation and differentiation. MAPK signalling regulated the proto-oncogene; activation of oncogenes by Cr6+ would support carcinogenesis.
In pathways of cellular energy metabolism and biosynthesis, a great majority of genes were found to be dysregulated. In energy metabolism pathway (Table VI), the upregulated genes would facilitate proton pumps for ATP synthesis, osmoregulation and active transport of molecules across cell membrane, nerve/muscle electrical excitability, and bioenergetics regulation. In biosynthesis pathway (Table VII), the upregulated genes would aid proteasomal degradation, biosynthesis dependent programmed cell function, and cell matrix homoeostasis.


In summary, these studies revealed that 300μM Cr6+ influenced the global cytogenomics and in particular regulated the stress, DNA repair, signalling system (Ca++, G-protein), Src kinase, MAPK and CDK, oncogenes, bioenergetics, and cell cycle. Specificity of oncogene expression to Cr6+ exposure may be predisposed by the adenocarcinomas cell line based test system.
BEAS-2B cells, 10 μM sodium dichromate, 4 h exposure: Andrew et al83 investigated the cellular gene expression profile of human bronchial epithelial BEAS-2B cells using a limited microarray of 1200 genes. They examined changes in gene expression after acute exposure to 10 μM dose of the toxicant. Cr6+ was found to modulate a cluster of 44 genes (Tables I-V).
In stress pathway (Table I), none of the genes showed upregulation; only heat shock protein genes (hsp40, hsp60, hsp90) recorded downregulation. The molecular basis seemed to be the protein transport and chaperon like functions. Their downregulation indicated irregular trafficking of deformed proteins and the risk of apoptosis.
Downregulation of anti-apoptotic proteins (Table II), DIF-2, was also in line with above observation. Similarly, downregulation of anti-death protein ephrin type-A receptor 2 ECK (tyrosine kinase receptor) demonstrated predisposition to apoptosis.
The trend of apoptosis was also noticed by the downregulation of genes in cell cycle pathway (Table III). ERF1 (EGF response factor-1) encoded the transcriptional activator C2H2-type zinc-finger nuclear protein that was functionally tumour suppressor and important for cell growth and differentiation. Cyclin K protein regulated cyclin dependent kinase and RNA polymerase. LUCA 2 (lysosomal hyaluromidase 2) transcribed the hyaluronidase, a cell surface protein, associated with tumour suppression function and involved in cell growth, differentiation and migration. For cell cycle regulation in Cr6+ exposed cells, TNK1 (tysosine kinase 1) encoded the non-receptor type of tyrosine kinase that phosphorylated proteins downstream of Src kinase in intracellular signalling (Table III). YWHA1 (tyrozine 3-monoxygenase / tryptophan 5-monooxygenase activation protein) encoded the signal transducers; FGFR1 (fibroblast growth factor receptor 1) encoded the fibroblast growth factor receptor type of protein to modulate growth and differentiation. Their underexpression seemed to be in consonance with a shift of cell cycle into apoptosis mode or the dedifferentiation mode.
Cr6+ induced predisposition of cells to apoptosis or transformation in tumour phenotype was perceptible by the sluggish DNA repair and metabolism (Table IV) through downregulation of genes involved in DNA template elongation, DNA binding and replication, G0 to G1/S phase of cell cycle, the cell growth, senescence, and differentiation. In DNA repair group of genes, only HATB2 (histone acetyl-transferase B subunit) (aka RBBP7/ RBBP46) was upregulated; it was functionally involved, as histone acetyltransferase, to import the acetylated histone into nucleus and deposit on up-coming DNA chains for chromatin assembly. Its overexpression would support DNA synthesis and possibly also the cell growth and differentiation.
Among oncogenes (Table V), c-myc (myelocytomatosis), FRA1 (Fos like antigen 1), PKB/akt (murine thymone viral ancogene), PP-1a (protein phosphatise 1) and TNK1 (tyrosine kinase 1) were found to be downregulated. However, MAPKAP kinase was found to be upregulated. This protein helped in protein transport; its upregulation favoured transition of cells into S phase. The underexpression of c-myc (the transcription factor) seemed to down play the cell cycle progression and thus favoured apoptosis. Its erratic transcription is described in haematopoietic tumourigenesis9091. Gene FRA1 encoded the fos-like antigen. This protein zipped up with JUN proteins to form AP-1 transcription factor for regulation of cell proliferation, differentiation, and transformation. Downregulated PKB (AKT1) was a Ser/Thr protein kinase. Usually inactive in G0 phase of cell cycle; it is activated by (platelet derived growth factor) PDGF through phosphatidylinositol 3-kinase and in this form it negatively regulated apoptosis by phosphorylation of the participating proteins. The downregulated TNK1 catalyzed phosphorylation of proteins downstream the membrane bound kinase. It is involved in negative regulation of cell growth and has tumour suppressor function.
Cr6+ exposure influenced the genes84 controlling cytoskeleton and cell junction boxes through adhesion/extracellular matrix. All the examined genes were downregulated. COLα2 (collagen α2) encoded pro-collagen to maintain the matrix integrity. CTNNA1 (catenin alpha 1) encoded the cadherins that in association with actin participated in cell differentiation. ITGB4 (integrin beta 4) encoded integrins to form cell-matrix or cell-cell adhesions. These proteins regulated cell integrity and shape. In aberrant expression state, integrins are key players in invasive carcinomas. Gene ZYX (zinc binding protein) encoded zyxin (the zinc binding protein) that accumulated in phosphorylated form at the cell surface making focal adhesion sites along the actin cytoskeleton and thus participating in signal transduction mechanisms. UPAR is a urokinase type plasminogen activator which regulates migration and invasion of cells and CI-B18 gene participates in cell adhesion process. Low expression of this gene could dysregulate the adhesion-molecule dependent changes favouring cell transformation.
The examination of genes regulating energy metabolism (Table VI) revealed the increased expression of glucose transport gene GLUT1 in Cr6+ exposed cells; it appeared to support major transport of aldoses including pentoses. A1ATR encoded the serine protease inhibitor that controlled inflammation; its downregulation would support inflammation essential for Cr6+ toxicity. Commensurate with apoptosis, downregulation of C1-B18 indicated the loss of cell differentiation potential and bioenergetics to regulate S phase of cell cycle.
Cr6+ exposure was shown to result in downregulation of several transcription regulators and suppressors (Table VII). A tone-down in level of these proteins could affect the S phase of the cell cycle requiring biosynthesis of proteins and thus influence cell proliferation and differentiation. This study83 suggested again the role of test dose-linked threshold switch which is crucial for Cr6+ toxicity in cells. However, a paucity of information on the observed cytogenomics vis-a-vis toxicity was notable. Cr6+ induced changes in transcription regulators and suppressors seen in the test system that lacks functional p53 and Rb gene showed the responsiveness of collateral genes and not the target gene to the toxicant.
BEAS-2B cells, 0.25 & 0. 5 μM potassium dichromate, 4 week exposure: In BEAS-2B cells, Sun et al84 for the first time reported the altered gene expression profile with respect to Cr6+ toxicity. They established Cr6+ transformed cell lines using 0.25 or 0.5 μM dose and 4-week long exposure condition and investigated the gene expression profile; the experimental conditions simulated the occupational exposure and the associated lung cancer. Microarray analysis using 28,869 gene array revealed the differential expression of genes recording >1.5 fold change in >1200 genes. A major group of genes was found to be commonly dysregulated in 0.25 or 0. 5μM Cr6+ transformed cell; and functionally associated to biosynthesis, apoptosis, cell junction desmocollin 2 (DSC2), desmocollin 3 (DSC3), prolyl endopeptidase (Prep), extracellular matrix ADAM (ADAM metallopeptidase domain 12), TIMP metallopeptiase inhibitor (TIMP3), matrix metalloproteinase-2 (MMP2), cysteine-rich secretory protein (CRISPLD2), cell adhesion (CDH6), cllaudin 1 (CLDN1), L1 cell adhesion molecule (LICAM), latrophilin 2 (LPHN2), absent in melanoma (AIM1), integrin alpha (ITGA), collagen type 4 alpha 1 (COL4A1), (COL5A1,2) and biglycan (BGN), laminin beta 1, gamma 2 (LAMB1, LAMC2), fibrullin (FBLN1, FBLN2). Mostly upregulated genes associated with cell junction, cell adhesion and extracellular matrix in Cr6+ transformed cells. Four genes namely latrophilin 2 (LPHN2), absent in melanoma 1 (AIM1), magtrix metallo peptidase 2 (MMP2), and cysteine-rich secretory protein LCCL domain containing 2 (CRISPLD2) were downregulated. Downregulated genes signified acquisition of carcinoma phenotype in Cr6+ exposed cells. Upregulation of cyclins in Cr6+ transformed cells in contrast to downregulation of TGFβ signalling system seemed to support cell transformation. Downregulated genes were related to integrins, collagens, laminin, and fibrullin components. HHIP (hedgehog interacting porotein) gene which antagonized hedgehog signalling pathways was underexpressed in these transformed cells. Study revealed the dysregulation of genes associated with cell adhesion, integrin receptor, cell matrix component, metalloproteinase indicating the loss of cell contact inhibition process of normal cells which is a cardinal change in cell transformation.
BJ-hERT cells, 0-6, 9μM sodium chromate tetrahydrate, 4-24 h exposure: In this study, researchers developed a sub-population of telomerase-transfected human fibroblast (BJ-hTERT) called as B-5Cr which survived the lethal dose of Cr6+. These transgenic and apoptosis resistant cells had an increased growth potential85. A genotoxic dose of 0-6, 9 μM Cr6+ induced apoptosis in BJ-hERT cells but B-5Cr cells, that were resistant to apoptotic dose of 0-6, 9 μM Cr6+, ignored the apoptotic signal of secondary Cr6+ insult. In order to investigate the molecular basis of such a selective clonogenic cell survival response to Cr6+ , the analysis of gene expression was performed after exposure to secondary doses (0-6 and 9μM) of Cr6+ in B-5Cr and BJ-hTERT cells using human genome arrays. In apoptosis resistant transgenic B-5Cr cells, results revealed dysregulation of genes involved in cell cycle regulation, and apoptosis besides the dysregulation of genes in DNA repair irrespective of toxicant exposure period (Table IV). Cell cycle regulatory gene p21 (WAF1, i.e. cyclin-dependent kinase inhibitor-1) was upregulated in both cell populations. Those genes that up-regulated in BJ-hTERT but not in B-5Cr were the genes of pathways regulating growth arrest, DNA-damage-inducible and apoptosis involving GADD45, Caspase 3, MKP5, Myc, c-rel oncogene, VDAC (voltage dependent ion channel) gene. Genes that upregulated only in B-5Cr cells and not in BJ-hTERT cells included DNA repair endonuclease gene XPF (xeroderma pigmentosum group F), Collagenase type 4, Bcl-xL, and ligand induced apoptosis signalling receptor DR6. UV-RAG gene was upregulated in B-5Cr cells85. Pritchard et al (2005)85 revealed that most of the genes altered by Cr6+ in transgenic cells were involved in apoptosis, cell cycle and DNA repair pathways. The upregulation of GADD45, caspase 3 in BJ-hTERT cells and bcl-XL in B-5cr cells demonstrated the molecular basis for acquisition of apoptosis or clonogenic transformation potential following Cr6+ exposure in respective type of cells85. Upregulation of DNA repair gene in transgenic B-5cr cells indicated the possibility of clonogenic transformation of cells after DNA damage. The study85 suggested that cells with competent or upregulated DNA repair mechanism can withstand apoptosis stimulus. Together with ‘adequate-to-survive DNA repair’ status, clonal expansion of cells can occur; these cells can escape the cell death signal in presence of the upregulated DNA repair mechanism and can thus be selected clonogenically for tumourigenesis.
Human PBMC cells, 0.2-10 μM sodium dichromate, 18 h exposure: In a study to identify biomarkers for Cr6+ exposure using human PBMC, change in gene expression was probed as the early markers using human gene chip of 18,000 transcripts86. Researchers selected PBMC as the test system and test dose of 0.2 μM Cr6+ to analyze gene expression alteration in immunoregulatory pathways. The test dose was biologically effective as it decreased chemokine secretion; the dose of 0.2 μM was preferred over 10 μM dose that elicited an increase in chemokine secretion and was comparatively higher in view of the investigators. A cluster of 1,659 genes was found to be significantly altered by 0.2 μM Cr6. Genes pertaining to pathways of apoptosis, cell cycle regulation, and immune systems were found to be dysregulated (Tables II, III, VIII). Low expression of many immunoregulatory genes like CD163, MRC, CD93, CD14, SLAMF8, and STAB1 was noticeable (Table VIII). Several pro-apoptotic genes cell death protein gene R1PK1, apoptosis inducing serine/threonine protein kinase STK17B, death inducer obliterator DIDO1 were upregulated and the anti-apoptotic gene BIRC1 was downregulated indicating predisposition to apoptosis after Cr6+ exposure (Table II). The upregulation of cyclins (also seen in humans45), CDC proteins, cyclin dependent kinases, G2/M transition regulatory protein, and downregulation of growth arrest proteins supported observation of the role of thresholds in Cr6+ toxicity (Table III). The downregulation of genes encoding protein involved in vital functions like anti-inflammation process revealed the onset of inflammation after Cr6+ exposure, which strengthened the process of Cr6+ programmed cell death. The study86 indicated the modulation of many vital pathways by Cr6+ including - the immune response, intracellular signalling, apoptosis, along with the cellular metabolism, RNA transport and binding, biogenesis and organelle organization, and transition metal binding. Cr6+ induced changes seen in immunoregulatory gene expression may be non specific to toxicant as the test system was immune cell.

Human fibroblast cells, 5μM Potassium dichromate, 16h exposure: Investigations, using human dermal fibroblast cells and human Ref-8V2 Sentrix bead chip array87, revealed the global gene expression profile of Cr6+ exposed cells. Relation between dermatitis and Cr6+ is well known but the gene expression study in this aspect was done for the first time by Sellamuthu et al87. Dermal fibroblasts were cultured and exposed to 5μM (LC50 value) Cr6+ concentration. Total RNA was isolated for microarray study. Several apoptosis linked genes involved in p53 signalling pathways were found to be overexpressed, suggesting that apoptosis was p53 dependent. The pro-apoptotic genes were noted to be upregulated and the anti-apoptotic genes downregulated. A cluster of 1153 genes was found to be significantly altered (>1.8 fold). More than 200 dysregulated genes showed relation to the programmed cell death. Besides apoptosis, differentially expressed genes also belonged to cell death, cell viability and survival. This study emphasized on genes involved in apoptosis. Interestingly 300 genes participating in cancer pathway were found to be differentially expressed. Apart from cancer, potential of Cr6+ to impact pathways of inflammation, immuno-regulatory system, endocrine system, metabolism, and genetic disorder of skin was also noticeable; genes were found to be involved in cell function of growth and differentiation, signalling mechanism, transport, cell cycle regulation, protein metabolism, and cell development. These results validated the development of threshold for apoptosis after Cr6+ exposure in human fibroblasts.
In vivo study
Sprague-Dawley rats, 0.25 mg/kg b. wt., sodium dichromate for 3 consecutive days: Izzotti et al88 conducted a study on Sprague-Dawley rats after administering sodium dichromate; test dose was administered intratracheally and repeatedly. Gene expression profile was investigated in rat liver and lung using 216 gene nylon arrays. A cluster of 56 genes was found to be upregulated 3-fold in lung compared to liver (Tables II-VI); none of the examined genes was downregulated. Biological annotation analysis of the upregulated genes revealed their roles in pathways like stress response, DNA repair and metabolism, energy metabolism, biosynthesis, apoptosis, oncogene and cell cycle. In stress response genes (Table I), the observed upregulation in Cu/Zn SOD, HSP-70, and damaged protein degradation enzyme was similar to earlier observation82.
Amongst apoptosis related genes (Table II), overexpression of proteins catalyzing the pro-apoptotic activity was novel and included Bcl-XL, Bcl-2 associated death promoter, cyclins, CDC-like kinase, CDC phosphatase, cyclin dependent kinase regulators, microtubule constituents, Ser/Thr protein kinase specific to G1 to S phase transition of cell cycle. Proteins interacting with Rb gene product to allow mitosis, and protein kinase complex for progression of G1 phase of cell cycle were also overexpressed.
This study88 revealed upregulation of several genes encoding DNA metabolizing enzymes (Table IV) like DNase, topoisomerases, multifunctional DNA repair enzyme, DNA polymerase alpha/beta/delta1, telomerase associated protein. The changes seemed to be crucial for DNA repair process through endonucleolytic activity, changing topology of DNA, forming constituent of the ribonucleoprotein complex responsible for telomerase activity, and identification and repair of apurinic/apyrimidinic sites.
Cr6+ upregulated two oncogenes - ras and c-jun terminal kinase gene (Table V), which encoded cell transforming proteins. Their increased levels could help in growth of transformed cells. The upregulation of genes involved in energy metabolism was also noted (Table VI). Their protein products (namely flavin containing monooxygenases, epoxide hydrolases, Cytochrome b5 reductase, acyl-coenzyme A dehydrogenase, aldehyde dehydrogenase, thiosulphate sulphurtransferase) played critical role in influencing the metabolism of xenobiotics, fatty acid (long chain and very long chain), cholesterol, various aldehydes, lipid peroxides, corticosteroids, neurotransmitters, and sulphur containing proteins. In summary, this study revealed that Cr6+ administration caused selective changes locally at the site of administration albeit similar as seen in cultured cells82. Altered gene expression was seen in Cr6+ metabolism, stress response, DNA repair, signalling pathways, apoptosis and cell cycle regulation. The study, although limited, was contributory in understanding the mechanisms of Cr6+ toxicity and suggested an involvement of thresholds in Cr6+ toxicity.
B6C3F1 mouse, (0, 0.3, 4, 14, 60, 170 or 520 mg/l) sodium dichromate dehydrate for 7 & 90 days: Recently, Kopec et al89 conducted the study on B6C3F1 female mice to investigate the key events of Cr6+ induced tumour formation in vivo. Dose dependent gene expression profile was examined after 7 and 90 days of regular exposure to Cr6+ in drinking water. Mouse intestinal epithelial gene expression was investigated using mouse 4X44K whole genome oligonucleotide microarray containing 21307 genes. After seven days, the differentially expressed genes exhibited the comparable expression profiles at ≤14 or ≥60 mg/l dose. A cluster of 6562 unique differentially expressed genes was identified having >1.5 fold change at one or more doses in duodenum at 8th day. Using the same data filtering criteria, cluster of 4448 unique differentially expressed genes in intestinal samples was noticed at 8th day, and clusters of 4630 and 4845 were detected in duodenum and jejunum, respectively at day 91. In long term exposure study, the differentially expressed genes exhibited the dissimilar expression profile. Genes in duodenum and jejunum, responding to range of Cr6+ concentrations (0.3-150 mg/l) participated in functions, e.g. immunoregulation, oxidative stress, cell cycle, growth, proliferation, DNA damage / repair: only the selected intestinal genes that were differentially expressed following exposure to 0.3-520 mg/l sodium dichromate dehydrate have been listed in Tables I, III, V, VIII. Activation of oxidative stress responsive genes MT2, MTF-1, Gpx (glutathione peroxidase) and Sod was seen; Ye and Shi82 also observed changes in similar genes. In DNA repair pathway, base and nucleotide excision repair gene Apex1 (base & nucleotide excision repair gene) Mlh1, Msh2 (Mut S protein homology 2), and Msh6 (Mut S protein homology 6). A comparison of changes in gene expression after 7 or 90 days of exposure to toxicant revealed overlaps of gene expressions. Taken together, the study showed that oxidative stress and the cytotoxicity were the early effects of Cr6+ exposure and that the differentially expressed genes were associated with oxidative stress, cell cycle and immuno-regulation pathways.
Summary and conclusion
It is apparent that exposure to Cr6+ results in dysregulated expression of a large group of genes; and the differences in gene identity are related to Cr6+ test doses / concentrations and test systems. Dysregulated genes are not associated with any specific pathway; however, these may participate in specific cellular function. A few studies848589 have revealed the pattern of dysregulation and intensity of changes suitable for apoptosis, cell transformation, or carcinogenicity. The dysregulated genes are uncommon however, the dysregulated pathways are common; and their functions support Cr6+ toxicity or in vitro cell transformation. These studies show a strong upregulation of genes with respect to stress response, energy metabolism, DNA repair, cell cycle regulation; a moderate upregulation of genes with respect to biosynthesis, oncogene, apoptosis; and a strong downregulation of genes linked to immunoregulation, Gap/tight junction, focal/cell adhesion, extracellular matrix, cytoskeleton pathways.
The commonly upregulated genes seen in microarray based studies are related to stress response (ATF, Cu/Zn SOD, GPX, MTF), apoptosis (caspase4, Bcl-xl, Tnfrs10), cell cycle regulation (Cyclin D1, 2, 3, Cyclin E & G), DNA repair (Apex, Gadd45a), oncogene (MAPKAP kinase) and biosynthesis (Ubiquitin). The studies on gene expression using assorted genes also show similar results such as overexpression of p533845464748495051697071838487, activation of oxidative stress responsive genes MTF-18289, induction of histone alkylation535483, upregulation of cyclins8645. The common genes and pathways dysregulated by Cr6+ exposure indicate the resultant dynamics of cytogenomics, its intensity, and the possible flow of key mechanistic events irrespective of the toxicant exposure conditions and the test systems to culminate rationally into the expressed biological / clinical effects. These commonly upregulated genes can serve as biomarkers for biomonitoring Cr6+ exposure; however, more studies using different doses and test systems are needed to validate these logical conclusions.
A critical role of cytogenomics, intensity of altered gene expression, and the flow of crucial mechanistic events in Cr toxicity has been shown by various studies. Thus, a hypothesis can be drawn that the cytogenomics profile and its intensity forms the sub-threshold or threshold level of gene expression to navigate Cr6+ toxicity as illustrated in the Figure. Sequentially, Cr6+ , after the cellular uptake, can undergo metabolic reduction causing ROS generation, DNA damage (Figure: route ‘1a’), and/or the altered gene expression (Figure: route ‘1b’) in exposed cells. In absence of ‘adequate DNA repair’ and the persistence of DNA damage, there could be limited options for the exposed cells. With ‘adequate-to-survive DNA repair’, cells either may restore the normal process of cell growth and differentiation (Figure: route ‘IIa’); or with ‘unrepaired or faultily repaired DNA, cells may proceed to toxicity like cytotoxicity, necrosis, apoptosis (Figure: route ‘IIb’) or to transformation into tumour phenotype (Figure: route ‘IIc’). Hypothetically, this may be a critical and decision making step by the altered transcription in Cr6+ exposed cell. It is hypothesised that the decision making step is governed by the threshold of those altered biochemical reactions or the related interactions catalyzed by abnormally expressed genes or by those respective abnormally functioning pathways which critically manage stress, DNA damage, apoptosis, cell-cycle regulation, cytoskeleton, cell morphology, energy metabolism, biosynthesis, oncogenes’ expression, immune system, bioenergetics; even the cross-talks of these dysregulated pathways can be crucial for the onset of toxicity. To conclude, the strong, moderate, or feeble intensity of dysregulation and reversibility of gene-expressions or pathways may depend upon several factors like dose and type (including speciation) of toxicant, duration of toxicants’ exposure, type of target cells, their niche microenvironment, and bioavailability of cellular antioxidants92. The resultant differential intensity of dysregulation may become the decision maker to pave way eventually either to opt for reversal to normal differentiation and growth, or to result in toxicity like dedifferentiation or apoptosis commensurate to the exposure conditions in exposed cells of tissues or organs. The hypothesis however, needs more investigations and validation in different test systems to elucidate the affiliation of the critical changes with Cr6+ toxicity.
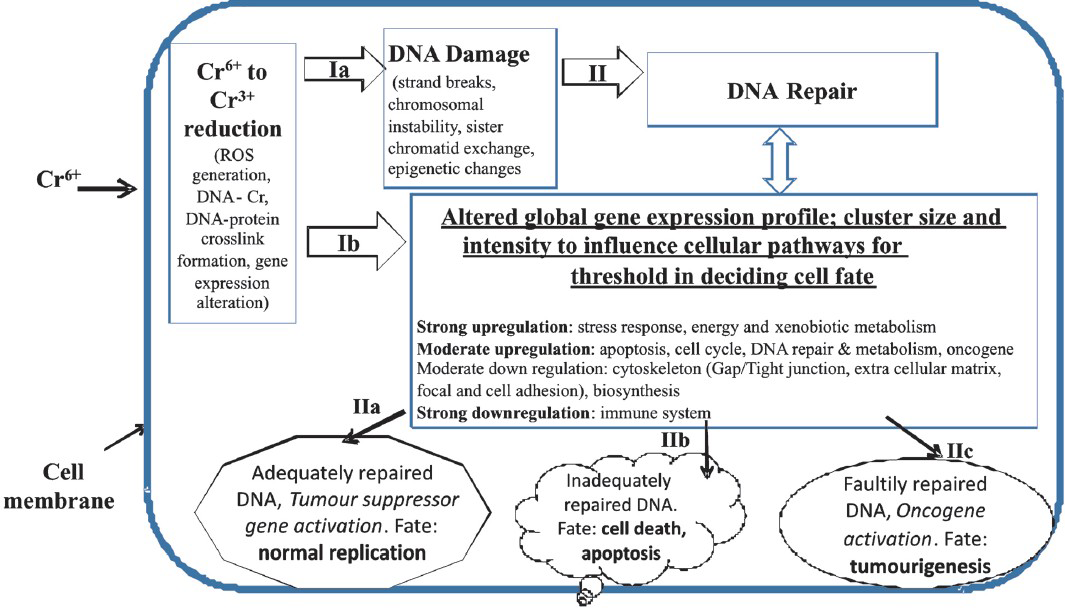
- Sequentially, Cr6+, after the cellular uptake, can undergo metabolic reduction causing ROS generation, DNA damage (via route ‘1a’), and/or the altered gene expression in exposed cells (via route ‘1b’). In absence of ‘adequate DNA repair’ and the persistence of DNA damage, there could be limited options for the exposed cells. With ‘adequate-to-survive DNA repair’ option, toxicant exposed cells may either restore the normal process of cell growth & differentiation (via route ‘IIa’); or with ‘unrepaired or faultily-repaired-DNA option, cells may proceed to toxicity like cytotoxicity, necrosis, apoptosis (via route ‘IIb’) or to transformation into tumour phenotype (via route ‘IIc’). Hypothetically, this is a critical and decision-making step in Cr6+ exposed cell for cell fate decisions that can be accomplished by the dose specific change in cytogenomics profile, gene expression intensity, status of DNA repair, and pathways for navigation of Cr6+ toxicity.
Acknowledgment
The authors acknowledge the Director, CSIR-Indian Institute of Toxicology Research, Lucknow, for encouraging the study, and the Indian Council of Medical Research, New Delhi, for providing research grant (IRIS 2005-00430) and to Council of Scientific and Industrial Research, New Delhi, for providing Senior Research Fellowship to the first two authors (AN, SP).
References
- Lung cancer mortality in nickel/chromium platers, 1946-95. Occup Environ Med. 1998;55:236-42.
- [Google Scholar]
- Occupational asthma induced by chromium salts. Allergol Immunopathol (Madr). 1989;17:133-6.
- [Google Scholar]
- One hundred years of chromium and cancer: a review of epidemiological evidence and selected case reports. Am J Ind Med. 1990;17:189-215.
- [Google Scholar]
- Cancer of the respiratory system in the United States chromate-producing industry. Public Health Rep. 1948;63:1114-27.
- [Google Scholar]
- Cytotoxicity of chromium-III and -VI compounds. 1. In vitro studies using different cell culture systems. Inhal Toxicol. 1993;5:345-69.
- [Google Scholar]
- Genotoxicity of chromium in human gastric mucosa cells and peripheral blood lymphocytes evaluated by the single cell gel electrophoresis (comet assay) Med Sci Monit. 2000;6:24-9.
- [Google Scholar]
- Genotoxicity and radioresistance in electroplating workers exposed to chromium. Mutat Res. 1999;446:23-34.
- [Google Scholar]
- The cytotoxicity and genotoxicity of particulate and soluble hexavalent chromium in human lung cells. Mutat Res. 2002;517:221-9.
- [Google Scholar]
- Particulate and soluble hexavalent chromium are cytotoxic and genotoxic to human lung epithelial cells. Mutat Res. 2006;610:2-7.
- [Google Scholar]
- Genotoxic evaluation of welders occupationally exposed to chromium and nickel using the Comet and micronucleus assays. Mutagenesis. 2004;19:35-41.
- [Google Scholar]
- Comparison of two particulate hexavalent chromium compounds: Barium chromate is more genotoxic than lead chromate in human lung cells. Environ Mol Mutagen. 2004;44:156-62.
- [Google Scholar]
- Neoplastic transformation of human bronchial cells by lead chromate particles. Am J Respir Cell Mol Biol. 2007;37:544-52.
- [Google Scholar]
- Characterization of chromium effects on a rat liver epithelial cell line and their relevance to in vitro transformation. Cancer Res. 1988;48:6484-90.
- [Google Scholar]
- Quantitative studies of in vitro morphological transformation of Syrian hamster cells by inorganic metal salts. Cancer Res. 1979;39:1008-13.
- [Google Scholar]
- In vitro transformation of BHK21 cells grown in the presence of calcium chromate. Cancer Res. 1975;35:1058-63.
- [Google Scholar]
- Arsenic and chromium enhance transformation of bovine papillomavirus DNA-transfected C3H/10T1/2 cells. Cancer Lett. 1996;103:65-9.
- [Google Scholar]
- Deficient repair of particulate hexavalent chromium-induced DNA double strand breaks leads to neoplastic transformation. Mutat Res. 2008;649:230-8.
- [Google Scholar]
- Chromosomal aberrations and morphological transformation in hamster embryonic cells treated with potassium dichromate in vitro. Mutat Res. 1977;46:87-94.
- [Google Scholar]
- Utilization of DNA-protein cross-links as a biomarker of chromium exposure. Environ Health Perspect. 1998;106(Suppl 4):969-74.
- [Google Scholar]
- Mechanism of DNA-protein cross-linking by chromium. Chem Res Toxicol. 2010;15(23):341-7.
- [Google Scholar]
- Chromium induces chromosomal instability, which is partly due to deregulation of BubR1 and Emi1, two APC/C inhibitors. Cell Cycle. 2011;10:2373-9.
- [Google Scholar]
- Microscopic analysis of the chromium content in the chromium-induced malignant and premalignant bronchial lesions of the rat. Environ Res. 2005;99:267-72.
- [Google Scholar]
- Ascorbate acts as a highly potent inducer of chromate mutagenesis and clastogenesis: linkage to DNA breaks in G2 phase by mismatch repair. Nucleic Acids Res. 2007;35:465-76.
- [Google Scholar]
- K-ras mutations in non-small-cell lung carcinoma: a review. Clin Lung Cancer. 2006;8:30-8.
- [Google Scholar]
- Aneugenic effects of some metal compounds assessed by chromosome counting in MRC-5 human cells. Mutat Res. 2000;469:35-40.
- [Google Scholar]
- Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium. Chem Res Toxicol. 2008;21:28-44.
- [Google Scholar]
- Epigenetic and gene expression changes related to transgenerational carcinogenesis. Mol Carcinog. 2004;40:1-11.
- [Google Scholar]
- Frequent microsatellite instability in lung cancer from chromate-exposed workers. Mol Carcinog. 2002;33:172-80.
- [Google Scholar]
- Effects of fume particles from stainless steel welding on sister chromatid exchanges and chromosome aberrations in cultured Chinese hamster cells. Ind Health. 1979;17:39-49.
- [Google Scholar]
- Sister chromatid exchange induced by chromium compounds in human lymphocytes. Int Arch Occup Environ Health. 1998;71:550-3.
- [Google Scholar]
- Sister chromatid exchanges induced in cultured mammalian cells by chromate. Chem Biol Interact. 1979;26:281-6.
- [Google Scholar]
- Chromosomal aberrations and sister chromatid exchanges of peripheral blood lymphocytes in Chinese electroplating workers: effect of nickel and chromium. J Trace Elem Exp Med. 1988;1:57-62.
- [Google Scholar]
- Sister-chromatid exchanges induced by some chromium compounds in human lymphocytes in vitro. Mutat Res. 1981;90:425-31.
- [Google Scholar]
- Differential DNA-protein crosslinking in lymphocytes and liver following chronic drinking water exposure of rats to potassium chromate. Toxicol Appl Pharmacol. 1991;109:60-72.
- [Google Scholar]
- DNA fragmentation, DNA-protein crosslinks, postlabeled nucleotidic modifications, and 8-hydroxy-2’-deoxyguanosine in the lung but not in the liver of rats receiving intratracheal instillations of chromium (VI). Chemoprevention by oral N-acetylcysteine. Mutat Res. 1998;400:233-44.
- [Google Scholar]
- Analysis of chromate-induced DNA-protein crosslinks with the comet assay. Mutat Res. 2000;471:71-80.
- [Google Scholar]
- Generation of S phase-dependent DNA double-strand breaks by Cr (VI) exposure: involvement of ATM in Cr (VI) induction of gamma-H2AX. Carcinogenesis. 2004;25:2265-74.
- [Google Scholar]
- Zinc chromate induces chromosome instability and DNA double strand breaks in human lung cells. Toxicol Appl Pharmacol. 2009;234:293-9.
- [Google Scholar]
- Occupational cancer genetics: infrequent ras oncogenes point mutations in lung cancer samples from chromate workers. Am J Ind Med. 2001;40:92-7.
- [Google Scholar]
- Malsegregation as a possible mechanism of aneuploidy induction by metal salts in MRC-5 human cells. Environ Mol Mutagen. 2002;40:200-6.
- [Google Scholar]
- The ATM-SMC1 pathway is essential for activation of the chromium (VI)-induced S-phase checkpoint. Mutat Res. 2004;554:241-51.
- [Google Scholar]
- Human bronchial epithelial cells malignantly transformed by hexavalent chromium exhibit an aneuploid phenotype but no microsatellite instability. Mutat Res. 2009;670:42-52.
- [Google Scholar]
- Frequent cyclin D1 expression in chromate-induced lung cancers. Hum Pathol. 2000;31:973-9.
- [Google Scholar]
- Mutations of the p53 gene in human lung cancer from chromate-exposed workers. Biochem Biophy Res Commun. 1997;239:95-100.
- [Google Scholar]
- Elevated serum levels of pantropic p53 proteins in chromium workers. Scand J Work Environ Health. 1997;23:37-40.
- [Google Scholar]
- Apoptosis and P53 induction in human lung fibroblasts exposed to chromium (VI): effect of ascorbate and tocopherol. Toxicol Sci. 2000;55:60-8.
- [Google Scholar]
- Chromium (VI)-induced oxidative stress, apoptotic cell death and modulation of p53 tumor suppressor gene. Mol Cell Biochem. 2001;222:149-58.
- [Google Scholar]
- Cytotoxicity and oxidative mechanisms of different forms of chromium. Toxicology. 2002;180:5-22.
- [Google Scholar]
- Long-term exposure to hexavalent chromium inhibits expression of tumor suppressor genes in cultured cells and in mice. J Trace Elem Med Biol. 2012;26:188-91.
- [Google Scholar]
- Chromium cross-links histone deacetylase 1-DNA methyltransferase 1 complexes to chromatin, inhibiting histone-remodeling marks critical for transcriptional activation. Mol Cell Biol. 2007;27:7089-101.
- [Google Scholar]
- Microsatellite instability and protein expression of the DNA mismatch repair gene, hMLH1, of lung cancer in chromate-exposed workers. Mol Carcinog. 2005;42:150-8.
- [Google Scholar]
- The reduced expression and aberrant methylation of p16 (INK4a) in chromate workers with lung cancer. Lung Cancer. 2006;53:295-302.
- [Google Scholar]
- Modulation of histone methylation and MLH1 gene silencing by hexavalent chromium. Toxicol Appl Pharmacol. 2009;237:258-66.
- [Google Scholar]
- Chromium (VI) inhibits the transcriptional activity of nuclear factor-kappaB by decreasing the interaction of p65 with cAMP-responsive element-binding protein-binding protein. J Biol Chem. 1999;274:36207-12.
- [Google Scholar]
- Molecular mechanisms of zinc-mediated induction and chromium (VI)-mediated inhibition of mouse Metallothionein-I gene transcription. J Health Sci. 2010;56:161-6.
- [Google Scholar]
- Chromate-induced epimutations in mammalian cells. Environ Health Perspect. 2002;110(Suppl 5):739-43.
- [Google Scholar]
- Chromium (VI) activates ataxia telangiectasia mutated (ATM) protein. Requirement of ATM for both apoptosis and recovery from terminal growth arrest. J Biol Chem. 2003;278:17885-94.
- [Google Scholar]
- Chronic exposure to particulate chromate induces spindle assembly checkpoint bypass in human lung cells. Chem Res Toxicol. 2006;19:1492-8.
- [Google Scholar]
- Cr(VI) increases tyrosine phosphorylation through reactive oxygen species-mediated reactions. Mol Cell Biochem. 2001;222:199-204.
- [Google Scholar]
- Activation of c-Jun N-terminal kinase in A549 lung carcinoma cells by sodium dichromate: role of dissociation of apoptosis signal regulating kinase-1 from its physiological inhibitor thioredoxin. Toxicology. 2004;197:101-12.
- [Google Scholar]
- Activation of MAP kinases by hexavalent chromium, manganese and nickel in human lung epithelial cells. Toxicol Lett. 2006;167:114-21.
- [Google Scholar]
- Activation of JNK, p38 and ERK mitogen-activated protein kinases by chromium (VI) is mediated through oxidative stress but does not affect cytotoxicity. Carcinogenesis. 2000;21:1491-500.
- [Google Scholar]
- Comparison of roles of three mitogen-activated protein kinases induced by chromium (VI) and cadmium in non-small-cell lung carcinoma cells. Mol Cell Biochem. 2001;222:85-95.
- [Google Scholar]
- Selective activation of Src family kinases and JNK by low levels of chromium (VI) Toxicol Appl Pharmacol. 2003;190:214-23.
- [Google Scholar]
- Distinct contributions of JNK and p38 to chromium cytotoxicity and inhibition of murine embryonic stem cell differentiation. Environ Health Perspect. 2009;117:1124-30.
- [Google Scholar]
- Cr (VI)-stimulated STAT3 tyrosine phosphorylation and nuclear translocation in human airway epithelial cells requires Lck. Biochem J. 2007;402:261-9.
- [Google Scholar]
- Participation of MAP kinase p38 and IkappaB kinase in chromium (VI)-induced NF-kappaB and AP-1 activation. J Environ Pathol Toxicol Oncol. 2000;19:231-8.
- [Google Scholar]
- On the mechanism of Cr (VI)-induced carcinogenesis: dose dependence of uptake and cellular responses. Mol Cell Biochem. 2001;222:221-9.
- [Google Scholar]
- Oxidative stress, hogg1 expression and NF-kappaB activity in cells exposed to low level chromium. J Occup Health. 2003;45:271-7.
- [Google Scholar]
- Hexavalent chromium Cr (VI) up-regulates COX-2 expression through an NFκB/c-Jun/AP-1-dependent pathway. Environ Health Perspect. 2012;120:547-53.
- [Google Scholar]
- Clinical implications for vascular endothelial growth factor in the lung: friend or foe? Respir Res. 2006;7:128.
- [Google Scholar]
- Vascular endothelial growth factor in the lung. Am J Physiol Lung Cell Mol Physiol. 2006;290:L209-21.
- [Google Scholar]
- Clinical significance of vascular endothelial growth factor in patients with primary lung cancer. Respirology. 2002;7:93-8.
- [Google Scholar]
- The prognostic significance of vascular endothelial growth factor levels in sera of non-small cell lung cancer patients. Respir Med. 2004;98:632-6.
- [Google Scholar]
- Alteration of protein expression pattern of vascular endothelial growth factor (VEGF) from soluble to cell-associated isoform during tumourigenesis. BMC Cancer. 2005;5:128.
- [Google Scholar]
- Signal transducer and activator of transcription 1 (STAT1) is essential for chromium silencing of gene induction in human airway epithelial cells. Toxicol Sci. 2009;110:212-23.
- [Google Scholar]
- Chromium (VI) stimulates Fyn to initiate innate immune gene induction in human airway epithelial cells. Chem Res Toxicol. 2010;23:396-404.
- [Google Scholar]
- AKT1 mediates bypass of the G1/S checkpoint after genotoxic stress in normal human cells. Cell Cycle. 2009;8:1589-602.
- [Google Scholar]
- Biological monitoring of hexavalent chromium and serum levels of the senescence biomarker apolipoprotein J / Clusterin in welders. Bioinorg Chem Appl. 2008;98:632-6.
- [Google Scholar]
- Gene expression profile in response to chromium-induced cell stress in A549 cells. Mol Cell Biochem. 2001;222:189-97.
- [Google Scholar]
- Genomic and proteomic profiling of responses to toxic metals in human lung cells. Environ Health Perspect. 2003;111:825-35.
- [Google Scholar]
- Comparison of gene expression profiles in chromate transformed BEAS-2B cells. PLoS One. 2011;6:e17982.
- [Google Scholar]
- Resistance to apoptosis, increased growth potential, and altered gene expression in cells that survived genotoxic hexavalent chromium [Cr(VI)] exposure. Mol Cell Biochem. 2005;279:169-81.
- [Google Scholar]
- Identification of human cell responses to hexavalent chromium. Environ Mol Mutagen. 2007;48:650-7.
- [Google Scholar]
- Transcriptomics evaluation of hexavalent chromium toxicity in human dermal fibroblasts. J Carcinog Mutag. 2011;2:1-8.
- [Google Scholar]
- Selective induction of gene expression in rat lung by hexavalent chromium. Mol Carcinog. 2002;35:75-84.
- [Google Scholar]
- Genome-wide gene expression effects in B6C3F1 mouse intestinal epithelia following 7 and 90 days of exposure to hexavalent chromium in drinking water. Toxicol Appl Pharmacol. 2012;259:13-26.
- [Google Scholar]
- Differential requirements for c-Myc in chronic hematopoietic hyperplasia and acute hematopoietic malignancies in Pten-null mice. Leukemia. 2011;25:1857-68.
- [Google Scholar]
- Lipoic acid prevents Cr6+ induced cell transformation and the associated genomic dysregulation. Environ Toxicol Pharmacol. 2013;36:182-93.
- [Google Scholar]




