Translate this page into:
Association of CFTR gene mutation with bronchial asthma
Reprint requests: Prof. Shally Awasthi, Department of Pediatrics, Chhatrapati Shahuji Maharaj Medical University, Lucknow 226 003, India e-mail: shallya@rediffmail.com
-
Accepted: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Mutation on both the copies of cystic fibrosis transmembrane conductance regulator (CFTR) gene results in cystic fibrosis (CF), which is a recessively transmitted genetic disorder. It is hypothesized that individuals heterozygous for CFTR gene mutation may develop obstructive pulmonary diseases like asthma. There is great heterogeneity in the phenotypic presentation and severity of CF lung disease. This could be due to genetic or environmental factors. Several modifier genes have been identified which may directly or indirectly interact with CFTR pathway and affect the severity of disease. This review article discusses the information related to the association of CFTR gene mutation with asthma. Association between CFTR gene mutation and asthma is still unclear. Report ranges from studies showing positive or protective association to those showing no association. Therefore, studies with sufficiently large sample size and detailed phenotype are required to define the potential contribution of CFTR in the pathogenesis of asthma.
Keywords
Asthma
CFTR
cystic fibrosis
heterozygous
modifier genes
phenotype

Introduction
Cystic fibrosis (CF) is the most common autosomal recessively transmitted genetic disorder among whites. Mutation on both the copies of cystic fibrosis transmembrane conductance regulator (CFTR) gene leads to CF. Dysfunction of CFTR Cl- channels in the genetic disease CF, disrupts transepithelial ion transport and hence the function of a variety of organs lined by epithelium1. This leads to the wide-ranging manifestations of the disease, which includes airway disease, pancreatic failure, meconium ileus, male infertility, and elevated levels of salt in sweat23.
The carriers of CFTR gene mutation are generally disease free but in some studies they have been found associated with pulmonary diseases like disseminated bronchiectasis (DB), allergic bronchopulmonary aspergillosis (ABPA), sinusitis, chronic obstructive pulmonary disease (COPD) and asthma4–8. It is not clear how CFTR alleles contribute to pulmonary disease, or whether the carrier status acts as a predisposing factor in conjunction with other genetic and environmental factors. In this review article we discuss all those studies which have assessed the association of CFTR gene mutation with asthma including the studies from India.
CFTR Gene
The CFTR gene was identified in 1989. It is a 230 kb gene on chromosome 7 encoding a 1480 amino acid polypeptide, named cystic fibrosis transmembrane conductance regulator (CFTR), which functions as a chloride channel in epithelial membrane910.
CFTR is a member of superfamily of adenosine triphosphate (ATP)-binding cassette (ABC) transporter ATPases, which requires energy of ATP hydrolysis to actively transport substrates across cell membrane. Among the thousands of ABC family members, only CFTR is an ion channel. CFTR is made up of five domains: two membrane-spanning domains (MSD1 and MSD2) that form the chloride ion channel, two nucleotide-binding domains (NBD1 and NBD2) that bind and hydrolyze ATP, and a regulatory (R) domain. While most ABC transporters consist of four domains (two membrane-spanning and two nucleotide-binding domains), CFTR is the only one known to possess a regulatory domain. Modification of the regulatory domain, either through the addition or removal of chemical phosphate groups, has been shown to regulate the movement of chloride ions across the membrane1112 (Fig. 1).
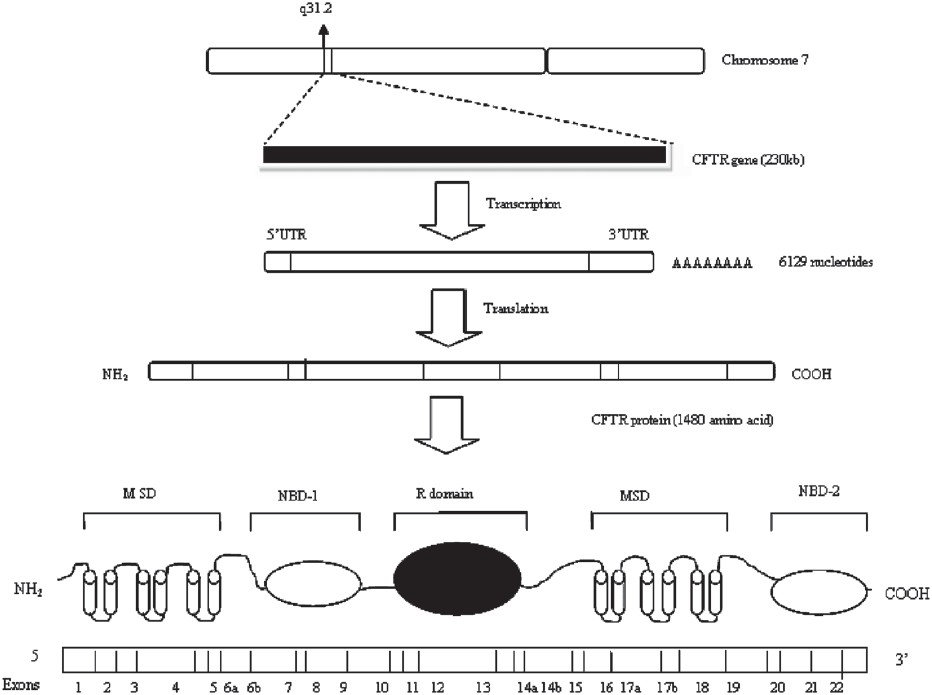
- Diagram showing CFTR gene and resulting protein (CFTR, cystic fibrosis transmembrane conductance regulator; MSD, membrane spanning domain; NBD, nucleotide binding domain; R domain, regulatory domain).
CFTR is located primarily in the apical membrane, where it provides a pathway for chloride ion (Cl–) movement across epithelium and regulates the rate of Cl– flow. In addition, CFTR regulates the activity of epithelial sodium channel (ENaC). In the sweat duct where electrolytes are reabsorbed, CFTR function is necessary for activating ENaC, whereas in respiratory epithelium where secretion and absorption occur, CFTR deregulates ENaC activity13. In the intestine, pancreas and sweat gland secretory coil, CFTR plays a key role in fluid and electrolyte secretion, whereas, in sweat gland duct and airway epithelia, it participates in fluid and electrolyte absorption14. Effects of the CFTR channel on vesicle trafficking, bicarbonate transport and the expression of inflammatory mediators such as Regulated upon Activation, Normal T-cell Expressed, and Secreted (RANTES), interleukin-8 (IL-8), interleukin-10 (IL-10) and inducible nitric oxide synthase (iNOS) have also been reported1516. Studies on CFTR expression show that it is also present in non-epithelial cell types including lymphocytes, cardiac ventricular cells, endothelial cells and more recently in rodent vascular and tracheal smooth muscle cells17–21.
CFTR gene mutations can be grouped into different classes based on their functional consequences on the CFTR within the cell. These mutations may be expressed in all domains of the protein10. Mutations in the CFTR gene are grouped into 5 classes: (i) Class I mutations (null mutations) do not produce the CFTR protein because of a premature stop signal in the CFTR DNA. (ii) Class II mutations do not interfere with the CFTR protein production, but the protein attains an unstable structure shortly after translation in the endoplasmic reticulum. (iii) Class III mutations result in the CFTR protein with reduced chloride transport on the basis of abnormal regulation of the chloride channel. (iv) Class IV mutations partially reduce chloride conductance through CFTRs. (v) Class V mutations lead to a severe reduction in the normal CFTR protein.
Classes I, II and III are severe and associated with the classic phenotype of sino-pulmonary disease and pancreatic insufficiency; IV and V are classified as mild and associated with variable phenotype, pancreatic sufficiency and borderline or normal sweat electrolyte concentration22.
Databases included
CFTR. OMIM: 602421; 219700 (CF); GDB: 120584; XM_004980; http://www.genet.sickkids.on.ca/cftr/ (CFTR Database; HGMD: CFTR).
The PubMed based comprehensive literature search was also done. The Medline database was used to gather initial sources for review. The key words used were “CFTR” OR “asthma” OR “CF heterozygosity” and “pulmonary function abnormality”. All articles identified by these search terms and related to the study were included for critical review.
Cystic fibrosis transmembrane conductance regulator: Genotypic and phenotypic spectrum
Genotype in CF is defined as the presence of mutation on each allele whereas, phenotype is an observable clinical features manifested in a patient, which is present as a combination of three variables: specific clinical components, their severity, and time course. The presence, severity and the time course of each clinical component may vary considerably among CF patients. Due to different clinical features of each affected organ, the relationship between genotype and each clinical component of the CF phenotype is evaluated separately23.
The most common mutation in the CFTR gene (responsible for 70% of abnormal CF gene) is a three-base-pair deletion that produces an inframe deletion of a phenylalanine (F) at position 508 (NBD1 domain). More than 1400 mutations reflecting all classes of genetic mutations have been identified in the CFTR gene to date (http://www.genet.sickkids.on.ca/cftr). These mutations may be expressed in all domains of the protein. The mutations affect CFTR through a variety of molecular mechanisms which can produce little or no functional CFTR at the apical membrane. This genotypic variation provides a rationale for phenotypic effects of the specific mutations. The extent to which various CFTR alleles contribute to clinical variation in CF is evaluated by genotype-phenotype studies. There is a broad phenotypic range between patients with both classic and mild variant CF, with high variability among patients carrying the same genotype and among siblings24. This variation may be because of both environmental factors and complex interaction between numerous gene products25. The degree of correlation between CFTR genotype and CF phenotype appears highest for pancreatic status and least for pulmonary disease23. Because of its complexity and patient exposure to a multitude of endogenous and exogenous factors, pulmonary outcome is clinically the most variable as well as the most unpredictable component of the CF phenotype. Atypical mild mutations in the CFTR gene have been linked to late onset pulmonary disease26. Several candidate genes related to innate and adaptive immune response have been implicated as pulmonary CF modifiers. The composition, frequency and type of CFTR mutations/variants parallel the spectrum of CFTR associated phenotypes, from classic CF to mild monosymptomatic presentations, such as various forms of obstructive azoospermia, idiopathic pancreatitis and disseminated bronchiectasis23.
Asthma pathogenesis
Asthma is a syndrome of variable airflow obstruction which has its primary origin in the airways that involves defective behaviour of the epithelium in relation to environmental exposures characterized pathologically by bronchial inflammation with prominent eosinophil infiltration and remodelling changes, physiologically by bronchial hyper-responsiveness, and clinically by cough, chest tightness and wheeze27.
In asthma the bronchial epithelium is highly abnormal, with structural changes involving separation of columnar cells from their basal attachments and functional changes including increased expression and release of proinflammatory cytokines (interleukins), growth factors (TGF) β 1,β2, and mediator-generating enzymes28. Environmental factors lead to reactivate the airways epithelial mesenchymal trophic unit (EMTU) that is involved in morphogenesis during foetal lung development29. Increased epithelial susceptibility and prolonged repair would serve to propagate the trophic response towards a chronic wound state in which mesenchymal cells (fibroblasts and smooth muscle cells) proliferate and alter their function to create the thickened and hyper-responsive airways characteristic of chronic disease30. The epithelial damage and TH2 cytokines act in together to cause a functional disturbance of EMTU which leads to remodelling ‘responses’ associated with bronchial hyperreactivity and asthma severity31.
Role of modifier genes in CF and asthma pathogenesis
Modifier genes are the genes other than primary disease causing gene, which alter the effects of the primary disease gene and, therefore, result in phenotypic variability. Interactions between CFTR genotype, modifier genes and environmental factors have been documented to influence lung function measures and infection status in CF patients32. These factors have led to the heterogeneity in the severity of the CF lung disease33. The general class of these potential modifier genes includes inflammatory and anti-inflammatory mediators, antioxidants, mediators of airway reactivity, molecules involved in CFTR trafficking, and alternative ion channels32. The best studied CF candidate modifiers include mannose-binding lectin (MBL), glutathione-S-transferase (GST), transforming growth factor-beta1 (TGF-B1), tumour necrosis factor-alpha (TNF-α), beta2-adrenergic receptor, and HLA class II antigens33. The individuals carrying the same genotype may present with the different phenotype because of the effect of these modifier genes. These genes (MBL, GST, TNF-α TGF-β1) which alter the CFTR pathway and affect the phenotype and severity of disease have also been found to be associated with inflammation and airway remodelling in asthma31–34. The individuals suffering from asthma and carrying the CFTR gene mutation only and those carrying CFTR mutation together with any of these modifier gene may have variation in the severity of the disease. These modifier genes can also effect the clinical presentation of the disease which may ultimately affect the treatment strategy. Therefore, studying gene modifiers will likely lead to a better understanding of pathophysiology and development of novel treatments.
Association of CFTR with asthma
The most common disease associated with CFTR, although incompletely defined is asthma. Till now several studies have been conducted in different populations and revealed interesting but conflicting facts about asthma development and CFTR gene variation, with some studies showing positive association of CFTR with asthma7835–37 and others showing either protective38 or no association39–45.
Beside the missense and deletion mutations in CFTR, some variants present at the polymorphic locus M470V and thymidine tract (Tn) of intron 8 have also been found to be associated with asthma35. The allele present at the polymorphic locus M470V affects the biogenesis of CFTR protein and the gating of the CFTR channel. The M470 protein has increased intrinsic chloride activity compared with V470 CFTR protein8. Polythymidine tract polymorphism is located in intron 8 near the acceptor site for exon 9. In normal population, three alleles are found at this locus (5Tat 0.05, 7T at 0.84 and 9Tat 0.11). Degree of exon skipping is inversely correlated with the length of Tn tract. Recent evidences have also shown that (TG) tract located immediately upstream of Tn tract can further modulate exon 9 skipping. These polymorphisms have been found responsible for alternative splicing of exon 9. CFTR mRNA without exon 9 produces a protein that is misfolded and nonfunctional. Thus carriers of 5T with exon 9 skipping have reduced channel activities and could have increased susceptibility for obstructive lung disease. These polyvariant alleles in combination with mutations, can affect the final phenotypic expression of the mutation46–48.
In 1995, first attempt to assess an association between asthma and the CFTR gene led to inconsistent results with a study, suggesting a protective association between heterozygosity for DF508 and asthma and another showing no association3839.
Dahl et al in 19987 conducted a study to find out the association between heterozygosity for the ΔF508 mutation and obstructive pulmonary diseases in a Danish population; 250 carriers (2.7%) of the ΔF508 mutation were identified, and of these 9 per cent carriers reported having asthma compared with 6 per cent of non carriers. They found a significant association between asthma and ΔF508 heterozygosity and concluded that ΔF508 heterozygosity is over-represented among asthmatics7 (Table I).

In the same year, Lowenfels et al40 conducted a multinational survey in 1113 obligate CFTR gene mutation heterozygotes and 688 controls and found that prevalence of asthma in CF heterozygotes was 9.6 per cent which was similar to that reported by Dahl et al40. A study from France reported that DF508 carriers were not at higher risk of any asthma-related phenotype41.
In 1999, Lazaro et al35 viewed the association by performing complete analysis of the CFTR gene in a sample of adult patients with asthma and in subjects from the general population and revealed 15 different missense mutations. Two control groups were included in the study in which control group 1 consisted of 41 samples from general population, spouses of CF carriers and were used as controls for the complete analysis of the CFTR coding region. Control group 2 consisted of 184 samples from anonymous blood donors and was used in the specific analysis of the recurrent variants found in the asthmatic group. Four of the 15 missense mutations (R75Q, G576A, R668C, and L997F) were detected in 57 per cent of the asthma patients. Missense mutations were not detected in the control group 1. However, these mutations were detected in control group 2 but the difference was not statistically significant. Only one subject with asthma had the common CF mutation ΔF508. They further identified five silent mutations in the coding region of CFTR (2377C/T, 2694T/G, 3690A/G, 4002A/G, and 4521G/A) and nine nucleotide changes in the non-coding region (125G/C, 406-6T/C, 712-93T/ A, 875+40A/G, 1398+13T/A, 1898+152T/A, 3601-65C/A, 3849+129delAT, and 4374+13A/ G) which were found in both control group 1 and the asthma patients at similar frequencies. Five new missense mutation (A534Q, V855I, T896I, M1028R, and T1142I) were identified in CFTR gene. None of the patients with missence mutation had 5T allele, while it was detected in 8 per cent of asthma patients without CFTR mutation and 9 per cent of subjects from general population. These findings suggested the role for combination of CFTR missence mutation, including M470V allele in genetic variability of asthma35 (Table I).
In a study on Greek population, the incidence of CFTR gene mutations and unclassified variants in chronic pulmonary disease such as asthma and other pulmonary diseases in children and adults was investigated and compared with subjects from general population8. Proportion of CFTR mutations was found 45 per cent in asthma, compared to 15.4 per cent in the general population. Seventeen different molecular defects involved in disease predisposition were identified in some cases, along with three potentially disease-causing mutations, T388 M, M1R and V11I, which were found only in three asthma patients. The IVS8-5T allele was found in two out of 20 asthma patients and one in control group whereas, proportion of M470V was 32.5 per cent in asthma patients compared to 35.5 per cent in control group. These results show the involvement of CFTR gene in asthma8 (Table I).
In French EGEA study 247 cases and 233 controls were compared42. Four most common mutations observed in Barcelona study (R75Q, G576A, R668C, and L997F) along with ΔF508, IVS8 and M470V were selected and studied. No significant association was found between the case and control group. ΔF508, 576A, 668C were significantly related to the M allele in M470V, whereas the opposite non-significant trend was observed for 75Q. The association of M470V with the other variants was similar in cases and controls42 (Table I).
Dahl et al44 conducted a study on adult Danish population. Individuals were genotyped to examine association between 5T, 7T, 9T and their 11 different genotype combinations, annual lung function decline and risk of asthma. They concluded that 5T heterozygosity was not associated with pulmonary dysfunction. They also found that ΔF508 heterozygosity was associated with increased asthma risk independently of the 5T allele36 (Table I).
In the Norwegian Environment and Childhood Asthma (ECA) study43, no significant association was found between asthma, reduced lung function, bronchial hyper responsiveness (BHR) or FeNO levels and CF heterozygosity (both when including or excluding the IVS8-5T polymorphism) or the modulating polymorphism [IVS8 (TG)mTn]. However, the IVS8 (TG) 11T7 haplotype alone was found associated with normal lung function43 (Table I).
In 2006, Ngiam et al37 investigated the potential relationship between CFTR gene mutation in Asian patients (Singaporean Chinese) with severe asthma and idiopathic bronchiectasis. Three missense mutations (1125T, 1556V, and Q1352H) and 1 splice variant (intron 8 12TG5T) were identified, representing a combined mutant/variant allele frequency of 0.25. Significantly higher frequency of CFTR mutations among patients with chronic pulmonary disease compared with unaffected controls suggests that these mutations may increase risk for disease37 (Table I).
A study was done in 2008 in Greek population to evaluate the association between CFTR gene mutations with asthma and pulmonary function abnormalities44. Study population comprised of 214 carriers and 185 non-carriers of CFTR gene mutations. It was found that the group of CFTR gene mutation carriers as well as the sub-group of ΔF508 carriers did not have any significant difference in prevalence of asthma compared to non carriers. However, according to pulmonary function testing, heterozygosity may be related with a silent obstructive pulmonary profile44.
In a case control study conducted in Korea45, 48 children with and without asthma were genotyped for the most common mutation (- 8G / C, Q98R, I125T, E217G, Q220X, A309A, M470V, I556V, T854T, Q1291X, Q1352H, R1453W) identified in Korean population along with variants in intron 8. No significant differences were found in genotype and allele frequencies of the 9 polymorphisms observed between the non-asthma and asthma groups. In a haplotype determination based on a Bayesian algorithm, 8 haplotypes were assembled in the 98 individuals tested and no any significant differences in haplotype frequencies between the non-asthma and asthma groups was observed45 (Table I).
Noone et al49 described two adult female patients with CF lung disease associated with the 5T allele to address the link between the 5T allele and disease at the molecular, cellular and organ level. They found that 5T polythymidine tract sequence on specific haplotype backgrounds (TG12 and M470V) may cause a low level of full length functional CFTR protein and CF-like lung disease49. However, other studies analyzing subjects with obstructive lung diseases have only focused on mutations that are known to cause CF.
Several studies done in context with asthma and CFTR gene mutation, have found increased IgE level and positive skin prick test among asthma patients. Association of ΔF508 heterozygosity to positive skin prick test of Aspergillus in a subsample of relatives and controls has been previously reported in a study41. However, a Spanish study found that the prevalence of positive skin test reactivity to Dermatophagoides pteronyssinus and the total IgE was lower in the group with missense mutations, but this difference was not statistically significant which suggests the tendency of the patients with CFTR mutations to be less allergic35. But these findings do not suggest the association of CFTR gene mutations with atopic asthma as most of the studies were done to find out the difference in the atopy-related parameters between the asthma and non-asthma groups.
Pathophysiology of asthma in relation with CFTR
CFTR dysfunction alters physiological functions of both surface and submucosal gland epithelium, which leads to salt and water imbalance across airway epithelium, depleted surface liquid layer, and impaired mucociliary clearance50. Impaired mucociliary clearance, together with CFTR-related changes in the airway surface microenvironment, leads to a progressive cycle of infection, inflammation, and declining lung function. Lung disease results from clogging of the airways due to mucus build-up, decreased mucociliary clearance and resulting inflammation. Inflammation and infection will cause injury and structural changes to the lungs. Airway surface liquid depletion is believed to cause ciliary collapse and loss of mucociliary clearance. Poor mucociliary clearance with excessive mucus production causes obstructive lung disease like asthma and chronic bacterial infections leading to respiratory failure which is major cause of mortality51–54 (Fig. 2).
- Pathophysiology of cystic fibrosis lung disease.
Cystic fibrosis asthma (CF asthma)
The term CF asthma refers to asthma like symptoms and bronchial hyper-responsiveness as have been reported in CF patients55. Both cystic fibrosis and asthma are characterized by airway inflammation and smooth muscle contraction due to stimulation by host inflammatory mediators56. There is no consensus on how to define CF asthma. It is difficult to determine who CF patients have asthma and who have asthma-like symptoms caused by CF lung inflammation. The diagnosis of asthma in CF patients is mainly clinical with several suggestive factors. Bronchial hyperreactivity and bronchodilator responsiveness are common in CF patients but a strong family and personal history of atopy may also point to asthma57.
CFTR in India
The incidence of CF in Caucasians is approximately 1 in 2500 children born in the United Kingdom58. It is less common in African Americans (1 in 15000) and in Asian Americans (1:31000). It also affects other ethnic groups such as black population with an incidence of 1 in 17,000 and the native American population with an approximate incidence of 1 in 80,00058. Initially the CF was thought to be rare in India that could be due to missed or under-diagnosed cases, but with advancement in diagnostic technologies it has become evident that CF does exist in India. Several studies have been conducted in different parts of India58–66 (Table II). ΔF508 is the most common mutation identified worldwide with a reported frequency of 66 per cent in CF patients66. However, the frequency of ΔF508 mutation in Indian children with classical CF is reported to be between 19-44 per cent5965. The spectrum of mutations other than ΔF508 in Indian patients is highly variable and some rare and new mutations have been observed61. Mutations 1161delC, 3849>10kbC-T and S549 N have been reported as other common mutations in Indian population beside ΔF5086061. Most of the studies have been done with respect to CF and Congenital bilateral absence of vas deferens (CBAVD)66. We could not come across any study assessing the association of CFTR gene mutation with asthma in Indian population.
Conclusion
At the molecular level CFTR heterozygotes almost certainly express the gene, though such expression may be masked or compensated by other genes and may be tissue specific. It has been shown that heterozygosity for cystic fibrosis is associated with increased airway reactivity and heterozygotes may be at risk for poor pulmonary function.
Heterozygotes with wheeze have been shown to be at higher risk for poor pulmonary function or development and progression of chronic obstructive lung disease. One gene for cystic fibrosis is sufficient to produce mild lung abnormalities in absence of infection. Study of heterozygotes may elucidate the pathogenesis of lung disease in cystic fibrosis. Studies on the expression of CFTR in the adult human lung have yielded conflicting results despite functional evidence of expression of CFTR in bronchiolar and alveolar epithelial cells. CFTR expression in alveoli needs to be investigated, in relation to bronchial asthma.
Although many studies in different population have been performed to find out the association of CFTR gene mutation with asthma, yet the results are inconclusive, as some of the studies have shown positive association while other could find either protective or no association. This can be due to difference in the ethnic groups selected for study or the influence of environmental factors on asthma pathogenesis besides the genetic predisposition. Therefore, further studies among different populations with asthma with well-defined criteria will help to verify these preliminary results and attempt to characterize a possible influence of CFTR gene mutation in aetiology of asthma disease.
References
- Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol Rev. 1999;79(Suppl 1):S77-107.
- [Google Scholar]
- Cystic fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular basis of inherited disease. New York: McGraw-Hill; 1995. p. :3799-876.
- [Google Scholar]
- Complete mutational screening of the CFTR gene in 120 patients with pulmonary disease. Hum Genet. 1998;103:718-22.
- [Google Scholar]
- CFTR gene mutations in adults with disseminated bronchiectasis. Eur J Hum Genet. 1997;5:149-55.
- [Google Scholar]
- Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations in allergic bronchopulmonary aspergillosis. Am J Hum Genet. 1996;59:45-51.
- [Google Scholar]
- Delta F508 heterozygosity in cystic fibrosis and susceptibility to asthma. Lancet. 1998;351:1911-3.
- [Google Scholar]
- CFTR gene mutations - including three novel nucleotide substitutions - and haplotype background in patients with asthma, disseminated bronchiectasis and chronic obstructive pulmonary disease. Hum Genet. 2001;108:216-21.
- [Google Scholar]
- Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;8:1066-73.
- [Google Scholar]
- Cystic Fibrosis and CFTR Gene. 2001. Atlas Genet Cytogenet Oncol Haematol. Available from: http://AtlasGeneticsOncology.org/Educ/CistFibID30032ES.html
- [Google Scholar]
- The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440:477-83.
- [Google Scholar]
- Update on pathogenesis of cystic fibrosis lung disease. Curr Opin Pulm Med. 2003;9:486-91.
- [Google Scholar]
- Characterization of CFTR expression and chloride channel activity in human endothelia. Am J Physiol. 1998;275:C1555-64.
- [Google Scholar]
- Regulation of the cystic fibrosis transmembrane conductance regulator channel by beta-adrenergic agonists and vasoactive intestinal peptide in rat smooth muscle cells and its role in vasorelaxation. J Biol Chem. 2004;279:21160-8.
- [Google Scholar]
- Disruption of CFTR chloride channel alters mechanical properties and cAMP-dependent Cl2 transport of aortic smooth muscle cells. J Physiol. 2005;568:483-95.
- [Google Scholar]
- Evidence that CFTR is expressed in rat tracheal smooth muscle cells and contributes to bronchodilation. Respir Res. 2006;7:113.
- [Google Scholar]
- Investigators of the ERCF.European Epidemiologic Registory of Cystic Fibrosis (ERCF): comparison of major disease manifestations between patients with different classes of mutations. Pediatr Pulmonol. 2001;31:1-12.
- [Google Scholar]
- Genotype-phenotype relationships in cystic fibrosis. Med Clin North Am. 2000;84:597-607.
- [Google Scholar]
- Pulmonary infection in mild variant cystic fibrosis: Implications for care. J Cyst Fibros. 2006;5:101-4.
- [Google Scholar]
- Cystic fibrosis 3849+10 KbC>T mutation associated with severe pulmonary disease and male fertility. Am J Respir Crit Care Med. 1996;153:858-60.
- [Google Scholar]
- Epithelial-mesenchymal interactions in the pathogenesis of asthma. J Allergy Clin Immunol. 2000;105:193-204.
- [Google Scholar]
- Airway remodelling in asthma - new insights. J Allergy Clin Immunol. 2003;111:215-22.
- [Google Scholar]
- Invited lecture: activation of the epithelial mesenchymal trophic unit in the pathogenesis of asthma. Int Arch Allergy Immunol. 2001;124:253-8.
- [Google Scholar]
- Cutting modifier genes in Mendelian disorders: the example of cystic fibrosis. Ann N Y Acad Sci. 2010;1214:57-69.
- [Google Scholar]
- Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am J Respir Crit Care Med. 1997;156:642-7.
- [Google Scholar]
- Mannose-binding lectin 2 (MBL2) gene polymorphism in asthma and atopy among adults. Clin Exp Immunol. 2005;142:120-4.
- [Google Scholar]
- Tumor necrosis factor-alpha: a promising therapeutic target for asthma? BioDrugs. 2007;21:345-9.
- [Google Scholar]
- Role of ADRB2 gene polymorphism in asthma and response to beta (2)-agonists in Polish children. J Appl Genet. 2009;50:275-81.
- [Google Scholar]
- Missense mutations in the cystic fibrosis gene in adult patients with asthma. Hum mutat. 1999;14:510-9.
- [Google Scholar]
- Asthma and COPD in cystic fibrosis intron-8 5T carriers.A population- based study. Respir Res. 2005;6:113.
- [Google Scholar]
- Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations in Asians with chronic pulmonary disease: A pilot study. J Cyst Fibros. 2006;5:159-64.
- [Google Scholar]
- Protection against bronchial asthma by CFTR DF508 mutation: a heterozygote advantage in cystic fibrosis. Nat Med. 1995;1:703-5.
- [Google Scholar]
- Heterozygotes for the delta DF508 cystic fibrosis allele are not protected against bronchial asthma. Nat Med. 1995;1:978-9.
- [Google Scholar]
- Delta F508 heterozygosity and asthma.EGEA Co-operative Group. Lancet. 1998;352:985-6.
- [Google Scholar]
- CFTR gene mutations and asthma in the Norwegian Environment and Childhood Asthma study. Respir Med. 2006;100:2121-2.
- [Google Scholar]
- Asthma and pulmonary function abnormalities in heterozygotes for cystic fibrosis transmembrane regulator gene mutations. Int J Clin Exp Med. 2008;1:345-9.
- [Google Scholar]
- Association between cystic fibrosis transmembrane conductance regulator gene mutations and susceptibility for childhood asthma in Korea. Yonsei Med J. 2010;51:912-7.
- [Google Scholar]
- Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nat Genet. 1993;3:151-6.
- [Google Scholar]
- Polyvariant mutant cystic fibrosis transmembrane conductance regulator genes.The polymorphic (TG) m locus explains the partial penetrance of the T5 polymorphism as a disease mutation. J Clin Invest. 1998;101:487-96.
- [Google Scholar]
- Functional analysis of cis-acting elements regulating the alternative splicing of human CFTR exon 9. Hum Mol. 1999;8:2339-49.
- [Google Scholar]
- Lung disease associated with the IVS8 5T allele of the CFTR Gene. Am J Respir Crit Care Med. 2000;162:1919-24.
- [Google Scholar]
- Evidence for airway surface dehydration as the initiating event in CF airway disease. J Intern Med. 2007;261:5-16.
- [Google Scholar]
- Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95:1005-15.
- [Google Scholar]
- Coordinated clearance of periciliary liquid and mucus from airway surfaces. J Clin Invest. 1998;102:1125-31.
- [Google Scholar]
- Mucociliary clearance in patients with cystic fibrosis and in normal subjects. Am J Respir Crit Care Med. 1994;150:66-71.
- [Google Scholar]
- Cystic fibrosis: Pathogenesis and future treatment strategies. Respir Care. 2009;54:595-602.
- [Google Scholar]
- International practice patterns by age and severity of lung disease in cystic fibrosis: data from the Epidemiologic Registry of Cystic Fibrosis (ERCF) Pediatr Pulmonol. 1997;24:147-54.
- [Google Scholar]
- Clinical profile and frequency of delta f508 mutation in Indian children with cystic fibrosis. Indian Pediatr. 2003;40:612-9.
- [Google Scholar]
- Delta F 508 molecular mutation in Indian children with cystic fibrosis. Indian J Med Res. 1996;104:355-8.
- [Google Scholar]
- Characterisation of mutations and genotype-phenotype correlation in cystic fibrosis: experience from India. J Cyst Fibros. 2008;7:110-5.
- [Google Scholar]
- Is the spectrum of mutations in Indian patients with cystic fibrosis different? Am J Med Genet. 2000;93:161-3.
- [Google Scholar]
- Carrier frequency of F508del mutation of cystic fibrosis in Indian population. J Cyst Fibros. 2003;5:43-6.
- [Google Scholar]
- Molecular diagnosis of cystic fibrosis in Indian patients - A preliminary report. J Assoc Physicians India. 2003;51:345-8.
- [Google Scholar]
- Application of multiplex ARMS and SSCP/HD analysis in molecular diagnosis of cystic fibrosis in Indian patients. Mol Diagn. 2005;9:59-66.
- [Google Scholar]
- Molecular basis of cystic fibrosis disease: An Indian perspective. Indian J Clin Biochem. 2010;25:335-41.
- [Google Scholar]




