
Translate this page into:
Association of BCL11A genetic variant (rs11886868) with severity in β-thalassaemia major & sickle cell anaemia
Reprint requests: Dr Anjana Munshi, Department of Molecular Biology, Institute Of Genetics & Hospital for Genetic Diseases, Begumpet, Hyderabad 500 016, Telangana, India e-mail: anjanadurani@yahoo.co.in
-
Received: ,
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
The amount of foetal haemoglobin that persists in adulthood affects the clinical severity of haemoglobinopathies including β-thalassaemia major and sickle cell anaemia (SCA). The present study was undertaken to analyse β-thalassaemia as well as SCA patients for the single nucleotide polymorphism (SNP), rs11886868 (T/C) in BCL11A gene and to evaluate the association between this polymorphism and severity of β-thalassaemia major and SCA.
Methods:
A total of 620 samples (420 β-thalassaemia major and 200 SCA cases) were analysed before blood transfusion using basic screening tests like complete blood analysis and osmotic fragility and further confirmed by high performance liquid chromatography (HPLC), amplification refractory mutation system-polymerase chain reaction (ARMS-PCR) and reverse dot blot techniques. All patients were transfusion dependent. Patients with β-thalassaemia and SCA were classified into mild, moderate, severe according to the severity score based on Hb levels, age of onset, age at which patients received their first blood transfusion, the degree of growth retardation and splenectomy. β-thalassaemia as well as SCA patients were analysed for the SNP, rs11886868 (T/C) in BCL11A gene and association between this polymorphism and severity of β-thalassaemia major as well as SCA was evaluated.
Results:
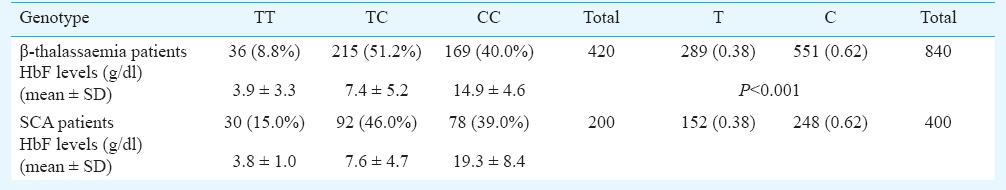
There was a significant difference in genotypic and allelic frequencies of BCL11A gene polymorphism between mild and moderate and mild and severe cases in both the groups. A significant (P<0.001) difference was observed in the mean HbF levels between the three genotypes in different severity groups. HbF levels were found to be high in CC genotype bearing individuals followed by TC and TT in β-thalassaemia major as well as SCA.
Interpretation & conclusions:
This study confirms that the T/C variant (rs11886868) of the BCL11A gene causing downregulation of BCL11A gene expression in adult erythroid precursors results in the induction of HbF and ameliorates the severity of β-thalassaemia as well as SCA.
Keywords
β-thalassaemia
erythroid precursors
foetal haemoglobin
sickle cell anaemia
single nucleotide polymorphisms (SNPs)

Clinical manifestations of β-thalassaemia and sickle cell anaemia (SCA) are extremely variable, ranging from nearly asymptomatic to severe and transfusion-dependant stage. The human β-globin gene cluster located on chromosome 11 has embryonic, foetal and adult globin genes, expressed sequentially during the various stages of development. The switch from human foetal (γ) to adult (β) globin gene expression has been of particular interest because of its clinical importance. Reactivation of foetal haemoglobin (HbF) or delay of switch in adult stage has been reported to ameliorate the clinical severity in β-haemoglobin disorders like β-thalassaemia and SCA1.
Genome wide association studies (GWAS) have identified genetic variants that fall outside of the β-haemoglobin gene. A new HbF associated locus on chromosome 2, located within the gene BCL11A (B-cell lymphoma/leukemia 11A) was identified by these studies12345. The BCL11A gene encoding a zinc finger transcription factor was shown to be downregulated in adult human erythroid precursor resulting in induction of HbF2.
In a previous GWAS in a Sardinian population the minor allele of single nucleotide polymorphism (SNP) rs11886868 in the BCL11A gene was found to associate significantly with an increase in the production of HbF6. However, an association could not be established between this SNP and increased HbF levels in some other ethnic groups56 indicating a discrepancy in different populations. Therefore, this study was carried out to investigate the frequency of SNP rs11886868 (T/C) in the BCL11A gene with severity of the β-thalassaemia major and SCA in patient population attending a tertiary care hospital in Telangana, India.
Material & Methods
The study was carried out at the Institute of Genetics and Hospital for Genetic Diseases, Hyderabad, Telangana, India. Of the 6500 individuals who visited this institute during January 2005 to December 2012, 420 patients confirmed to be affected with β-thalassaemia major and 200 with SCA were included in the study. Patients suffering from any other kind of haematological disorders were excluded from the study. Individuals who were carriers of the disease or had other structural variants of Hb were also excluded from the study.
Information on clinical characteristics of the patients was collected by using a structured questionnaire. Institutional Ethics Committee clearance was taken for the study. Blood samples (3 ml) were collected from the patients in EDTA vacutainers after obtaining written informed consent. Patients (n=620) confirmed to be affected by β-thalassaemia (n=420) or SCA (n=200) by high performance liquid chromatography (HPLC) and genomic DNA analysis were included in the study. All patients recruited in the study were transfusion dependent. HPLC technique was used with the β-Thalassaemia Short Program of Variant Bio-Rad (Bio-Rad, USA) (Figs 1, 2). The complete blood picture of all the patients was analysed before analysing the samples by HPLC.
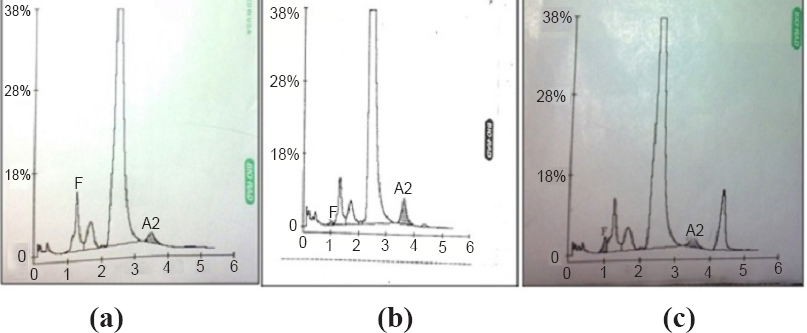
-
(a). HPLC chromatogram of a normal individual (A2- 2.1%; HbF-0.0% both A2 and HbF are in the normal range), (b) HPLC chromatogram of a β-thalassaemia carrier (A2-4.3%; HbF-0.4% A2 is high), (c) HPLC chromatogram of a sickle cell carrier (HbS- 8.8%; HbF-1.7% HbS in high range).
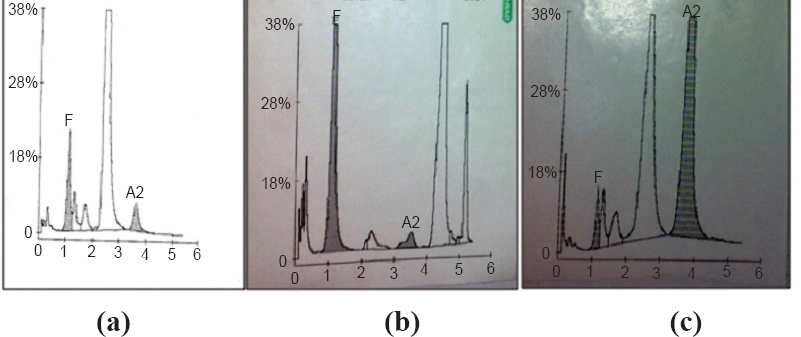
-
(a). HPLC chromatogram of a β-thalassaemia major (the chromatorgram is of a transfused beta thalassaemia major (A2-7.5%; HbF-3.7% both A2 and HbF are high), (b) HPLC chromatogram of a sickle cell anaemia (HbS-39.4%; HbF-10.6% both HbS and HbF are in high range), (c) HPLC chromatogram of a E-disease (A2-48.3%; hbF-3.9% A2 is too high HbF also high).
Clinical severity: The patients who had two β-thalassaemia alleles and two HbS alleles (based on mutation analysis) were categorized into mild, moderate, and severe groups by a systematic scoring system proposed by Sripichai et al7. Based on clinical observations alone, 310 patients were found to be thalassaemia intermedia (Hb level of at least 7g/dl at the time of diagnosis). Parameters that showed a correlation with disease severity (at r > 0.3) were included in the multiple logistic regression analysis. These were haemoglobin level, age of onset, age at which patients received their first blood transfusion, age at thalassaemia presentation, the degree of growth retardation, and splenectomy as reported previously89. The scoring system consisting of these six clinical criteria scored 0, 0.5, 1, or 2 according to clinical presentation. Patients whose severity score was <3.5, 3.5-7.5, and >7.5 were considered as mild, moderate, and severe cases, respectively.
Molecular studies: Genomic DNA was extracted from the whole blood samples using the phenol-chloroform method10. Amplification refractory mutation system (ARMS)-PCR and reverse dot blot (RDB) hybridization were used to screen the specific mutation in patients with β-thalassaemia and HbS in SCA as reported previously1112. Single nucleotide polymorphism of rs11886868 in the BCL11A gene (T→C) was detected by PCR-RFLP technique. A 548bp fragment was amplified using the primers 5’-TTTGGTGCTACCCTGAAAGAC-3’ and 5’-ACTCAACAGTAGCAGAATGAAAGAG-3 (http://simgene.com/Primer3). The 548-bp product was digested with MboII restriction enzyme (http://tools.neb.com/NEBcutter2/) by incubating at 37°C for five min, followed by separation of fragments on three per cent agarose gel. The T allele lacks the MboII restriction site, while it is present in the C allele. The C allele was detected as two fragments of 470 bp and 70 bp.
Statistical analysis: Association between the genotype and severity was examined by odds ratio with 95% confidence interval (CI) and Chi-squared analysis using Open Epi software (Department of Epidemiology, Rollins School of Public Health, Emory University, Atlanta, GA, USA). Allelic frequencies were calculated according to the number of alleles observed and the total number of alleles examined. The association of different genotypes with HbF percentage was evaluated by One-way ANOVA.
Results
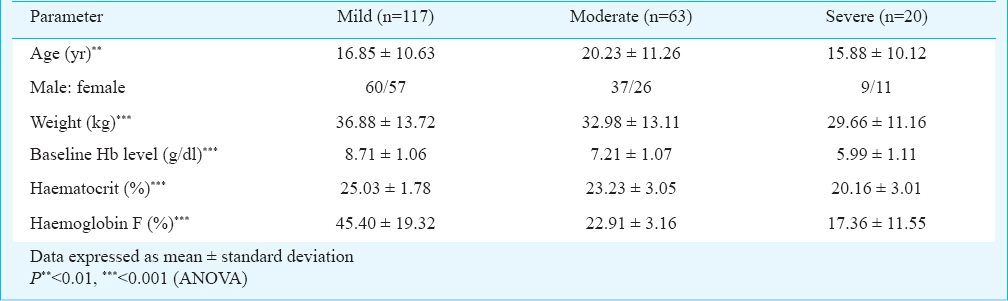
A total of 6500 individuals were tested using HPLC, ARMS-PCR (Figs 1–3) and RDB techniques for one of the heterogeneous group of congenital anaemia which includes β-thalassaemia, haemoglobin variants (D-thalassaemia, E-thalassaemia and SCA) and hereditary persistance of foetal haemoglobin (HPFH). The break-up of the analysed cases and the spectrum of mutations was in conformity with our previous publications89. The patients (420 β-thalassaemia, 200 SCA) were divided into subgroups (mild, moderate, and severe) based on the severity of the disease. The clinical data and biochemical characteristics of β-thalassaemia and SCA patients are given in Tables I and II. There were 131 mild, 179 moderate and 110 severe β-thalassaemia patients. The SCA patients in mild, moderate and severe groups were 117, 63 and 20, respectively. Genotypic and allele frequencies of BCL11A (T/C) gene polymorphism in both the patient group are given in Table III. There was a significant difference in the mean HbF levels between the three genotypes in different severity groups (P<0.001) in both β-thalassaemia and SCA patients. HbF levels were found to be higher in individuals with CC genotype followed by TC and TT genotype in β-thalassaemia major as well as in SCA patients (Table III). There was a significant difference in the genotypic distribution and allele frequency between mild and moderate, mild and severe groups in β-thalassaemia major as well as in SCA patients. With regards to BCL11A gene polymorphism, there was a significant difference in the high and low percentage HbF levels between the three genotypes in both SCA and β-thalassaemia patients (Tables IV, V).
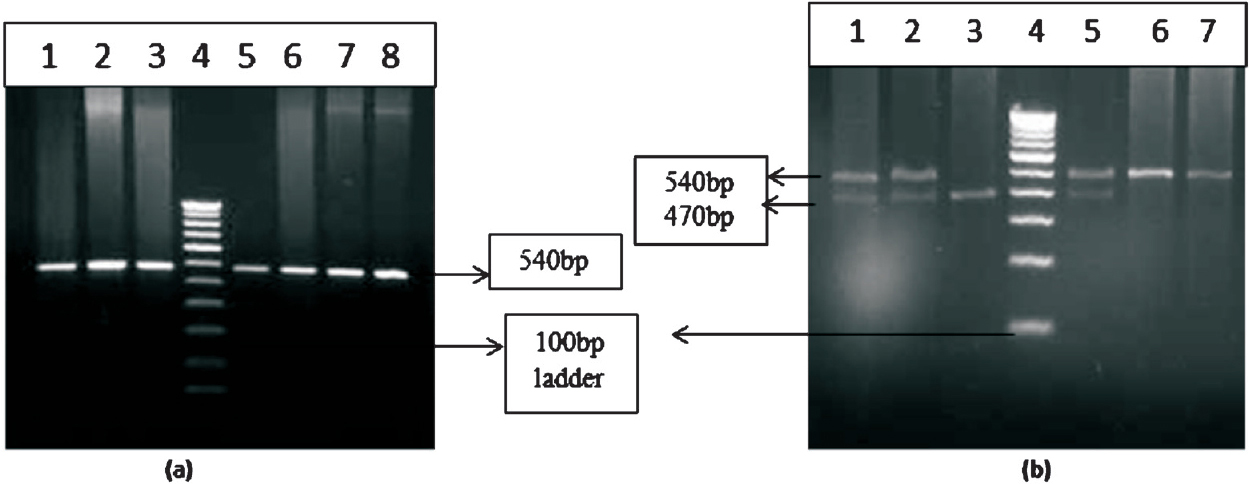
- Gel pictures of BCL11A gene polymorphism. (a) Lanes 1, 2, 3, 5, 6, 7 and 8 represent the amplified product of BCL11A gene and lane 4 represents 100 bp ladder. (b) restriction fragment length polymorphism (RFLP) of BCL11A gene polymorphism. Lanes 1, 2 and 5 represent heterozygous condition 6, and 7 represents homozygous condition. Lane 3 represents homozygous mutant.


Discussion
Modifying factors that reduce the imbalance of α- vs. Non α-haemoglobin chains are the determinants of clinical severity of β-thalassaemia as well as SCA. The reduced globin chain imbalance allows selective survival of the erythroid precursors and thereby reduces the unproductive erythropoiesis613.
It has been shown that the variants in BCL11A, HBS1L-MYB and HBB loci are associated with HbF levels and ameliorate the severity in β-thalassaemia as well as SCA1415. BCL11A has been described as a major suppressor of HbF expression. Several SNPs located in BCL11A gene and linked to HbF levels and thus the severity of β-thalassaemia and SCA have been described216. The direct binding of this transcription factor on to several locations in the β-globin cluster, especially to a region located at 5’to the δ-globin gene has also been demonstrated4. In a recent study it was found that the loss of this region was associated with an increase of 2.7g/dl of HbF. This would be sufficient to compensate the lack of Hb in β-thalassaemia major patients17.
The minor allele ‘C’ of SNP rs11886868 in the BCL11A gene has been shown to be a major modifier of HbF levels, than HBS1L-MYB locus or the co-inheritance of α-thalassaemia15. However, in Europeans HBS1L-MYB locus has been reported to have a stronger effect than BCL11A2. The different amount of variants explained by each marker reflects the heterogeneity of the allele frequencies among different ethnic groups. A significant association of CC genotype and C allele was observed with the milder disease phenotype in β-thalassaemia as well as SCA in the present study. The frequency of CC variant genotype was found to be 40 per cent in our study population. However, the frequency of the CC genotype has been found to be 28.8 per cent in an Iranian population18. Galanello et al15 have reported the frequency of CC genotype to be 61.2 per cent in Sardinians. The difference in the frequency of variant genotype might be on account of ethnic differences15. This study confirms that increased γ-globin expression associated with the BCL11A (T/C) polymorphism ameliorates the clinical severity in these haemoglobinopathies in the study population. The patients were also assessed based on the age at diagnosis; 70 per cent of the CC bearing individuals were diagnosed after one year of age, while 30 per cent were diagnosed at less than one year of age in β-thalassaemia, whereas in the SCA 76 per cent of CC bearing individuals were diagnosed after one year while 26 per cent were diagnosed at less than one year of age.
The role of BCL11A as a regulator of γ-globin gene silencing has also been demonstrated experimentally by increased production of HbF in developing adult erythroblasts after small hairpin RNA (sh-RNA) mediated knocked down19. BCL11A mediated silencing is orchestrated through cooperation with a high mobility group transcription factor SOX6, since BCL11A and SOX6 are co-expressed and interact physically1619. The data from genetic and functional studies support a key role for BCL11A in silencing of γ-globin genes during the developmental switching as well as its potential role in reactivation of HbF in adult erythroblasts.
In conclusion, the present study suggests that the HbF associated SNPs which increase the production of foetal Hb over the lifetime of a patient may be considered as an innate therapy for various haemoglobinopathies. Screening of BCL11A gene variants may eventually help to predict the severity of disease in newborn and will revolutionize the diagnosis, treatment and prevention.
Acknowledgment
The authors acknowledge the Department of Biotechnology (DBT), New Delhi, for financial support.
Conflicts of Interest: None.
References
- Discovering the genetics underlying foetal hemoglobin production in adults. Br J Haematol. 2009;145:455-67.
- [Google Scholar]
- A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p[15] Nat Genet. 2007;36:1197-9.
- [Google Scholar]
- The HBS1L-MYB intergenic region on chromosome 6q[23] is a quantitative trait locus controlling fetal hemoglobin level in carriers of β-thalassaemia. J Med Genet. 2008;45:745-51.
- [Google Scholar]
- Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc Natl Acad Sci USA. 2008;105:1620-5.
- [Google Scholar]
- Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839-42.
- [Google Scholar]
- Molecular analysis of β-zero-thalassemia intermedia in Sardinia. Blood. 1989;74:823-7.
- [Google Scholar]
- Scoring system for classification of β-thalassemia/HbE disease severity. Am J Hematol. 2008;83:482-4.
- [Google Scholar]
- Inherited hemoglobin disorders in Andhra Pradesh, India: A population Study. Clin Chim Acta. 2009;400:117-9.
- [Google Scholar]
- Lack of association of G779A ZHX-2 gene variant with HbF levels in β-thalassemia major. Euro J Haematol. 2011;86:502-6.
- [Google Scholar]
- Genotyping of BCL11A and HBS1L-MYB SNPs associated with fetal haemoglobin levels. BMC Genomics. 2014;15:108-20.
- [Google Scholar]
- Rapid and simultaneous typing of hemoglobin S, hemoglobin C, and seven Mediterranean beta-thalassemia mutations by covalent reverse dot-blot analysis: application to prenatal diagnosis in Sicily. Blood. 1993;81:239-42.
- [Google Scholar]
- A multi-center study in order to further define the molecular basis of β-thalassemia in Thailand, Pakistan, Sri Lanka, Mauritius, Syria, and India, and to develop a simple molecular diagnostic strategy by amplification refractory mutation system-polymerase chain reaction. Hemoglobin. 2001;25:397-407.
- [Google Scholar]
- The thalassemia syndromes. Oxford, United Kingdom: Blackwell Science; 2001. p. :704-12.
- DNA polymorphism at the BCL11A, HBS1L-MYB, and β-globin loci associates with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci USA. 2008;105:11869-74.
- [Google Scholar]
- Amelioration of Sardinian β0-thalassemia by genetic modifiers. Blood. 2009;114:3935-7.
- [Google Scholar]
- Estimation of the difference in HbF expression due to loss of the 5’ δ-globin BCL11A binding region. Haematology. 2013;98:305-8.
- [Google Scholar]
- The influence of the BCL11A polymorphism on the phenotype of patients with beta thalassemia could be affected by the beta globin locus control region and/or the Xmn1-HBG2 genotypic background. Blood Cells Mol Dis. 2013;51:80-4.
- [Google Scholar]
- Transcriptional regulation of fetal to adult hemoglobin switching: new therapeutic opportunities. Blood. 2011;117:3945-53.
- [Google Scholar]




