
Translate this page into:
Assessing oxidative stress in foetuses with β-globin gene mutations
For correspondence: Prof Pratima Kumari Sahu, Department of Biochemistry, SCB Medical College & Hospital, Cuttack 753 007, Odisha, India e-mail: drpratimasahu@gmail.com
-
Received: ,
Accepted: ,
Abstract
Background & objectives
Haemoglobinopathies, particularly thalassemia and sickle cell disease, are of major public health concern and pose a significant health burden in India particularly in the State of Odisha. The molecular complexity of β-thalassemia involves over 350 mutations, resulting in reduced beta-globin synthesis, excess iron, and oxidative stress.
Methods
Chorionic villi samples from β-thalassemia carrier mothers collected through trans-abdominal chorionic villi sampling (TA-CVS) were screened by real-time polymerase chain reaction for the most commonly found β-globin gene mutations: IVS 1-1 (G>T), 619 bp deletion, IVS 1-5 (G>C), CD15 (G>A), FS41/42 (-TTCT), HbS, FS8/9 (+G), -90 (C>T), CD16 (-C), HbE, CD15 (-T), CD30 (G>C), and -28 (A>G) using TOCE™ (Tagging Oligonucleotide Cleavage and Extension) technology. Reduced glutathione (GSH), redox status (GSH/GSSG ratio), and endothelin-1 (ET-1) were investigated in foetuses of defined β-globin gene mutations by colorimetry and ELISA, respectively, to explore the potential of GSH and ET-1 as oxidative stress biomarkers.
Results
Of the total cases included in this study 40 per cent showed occurrence of HbS mutations with significant differences in GSH, GSSG, and redox ratio among mutation groups (P<0.05). Correlation analysis revealed a non-significant association between GSH and ET-1 levels.
Interpretation & conclusions
This study provides key insights into oxidative stress in foetuses with defined β-globin gene mutations. GSH and ET-1 may be therapeutic targets to mitigate oxidative stress and healthy placentation in pregnancies with haemoglobinopathies.
Keywords
Beta-thalassemia
chorionic villi sampling
endothelin-1
oxidative stress
redox ratio
reduced glutathione
sickle cell disease

Thalassemia and sickle cell disease (SCD) are among the most prevalent chronic, life-restricting genetic disorders and need long-term, specialised treatment. These conditions not only cause immense physical and psychological distress but also result in a substantial economic burden on families with considerable strain on healthcare resources in India1.
The frequency of the sickle β-globin gene variant (βS) allele demonstrates its highest prevalence across Central India (up to 10%) in regions spanning southeastern Gujarat to southwestern Odisha. India has been ranked as the country with the second-highest number of predicted SCA births globally2. In 2019, both β-thalassemia and sickle cell hemoglobinopathy were reported to account for 50.2 per cent of anaemia cases in Odisha3. A two-year hospital-based study conducted in southern Odisha highlighted the spectrum of various abnormal haemoglobin variants among the patient population, providing valuable insights into the regional burden of these genetic conditions4.
The World Health Organization (WHO) report, 2008 States that approximately 1.1 per cent couples are at risk of having children with a haemoglobin disorder, and 2.7 per one thousand conceptions are affected worldwide5. Providing affordable therapy and reducing the number of affected children are two primary objectives of WHO towards addressing hemoglobinopathies6.
More than 350 β-thalassemia mutations have been reported to the Itha Genes database7. Primarily, the mutations are single nucleotide substitutions, deletions, or frameshift-inducing oligonucleotide insertions8. Specific β-globin gene mutations were selected as reported previously to capture most mutation types seen in this region basing on their high prevalence in the eastern Indian population, particularly Odisha9,10. While the βS variant is highly prevalent, its specific role in exacerbating oxidative stress biomarkers during pregnancy remains underexplored.
Oxidative stress, an imbalance between oxidant and antioxidant forces, plays a critical role in vascular pathways or perfusion of the placenta. It has a role in vascular endothelial growth factor (VEGF) and angiopoietin-1 pathways that influence downstream signalling and transcription factors vital for placental vascular development. Metabolomic studies on chorionic villi samples from β-thalassemia foetuses11 have highlighted the presence of oxidative stress and a metabolic shift toward the pentose phosphate pathway (PPP). This pathway is essential for maintaining reduced glutathione (GSH) levels to prevent oxidative damage in β-thalassemia fetuses11.
GSH and endothelin-1 (ET-1) in chorionic villi samples serve as critical defences against oxidative stress. These markers prove the importance of autophagy and apoptosis, two interlinked processes in the placenta that are significantly influenced by oxidative stress. GSH participates in diverse cellular processes, including protein synthesis, DNA repair, enzyme activity, metabolism, gene expression, and intracellular signaling12-15.
Endothelins (ETs), a family of 21-amino acid cysteine-rich peptides, are synthesised by endothelial and smooth muscle cells. ET-1, the most studied member, binds to G-protein-coupled Endothelin-1 receptor A (ETA) and Endothelin-1 receptor B (ETB), contributing to systemic and pulmonary artery resistance16. Therapeutic interventions targeting ET-1 receptors, including selective ETA and dual ETA/ETB antagonists, have shown potential in managing conditions associated with ET-1 overexpression. In the placenta, ET-1 plays a pivotal role in regulating uteroplacental circulation and facilitating the closure of the ductus arteriosus. However, its dysregulation can contribute to hypoxia-induced complications, exacerbating haemolysis of sickle-shaped red blood cells and amplifying the interaction between hypoxia, oxidative stress, and thrombosis in sickle cell disease16-18.
The present study aims to elucidate the interplay between different spectra of β-globin gene mutations and oxidative stress biomarkers in foetuses with hemoglobinopathies, which may influence foetal outcomes.
Materials & Methods
The study was conducted at the tertiary clinical care centre in Sriram Chandra Bhanj Medical College and Hospital (SCB MCH), Cuttack, Odisha, India after approval of the Institutional Ethical Committee from August 2023 to June 2024. The sample size was calculated using the G*Power software with a 0.8 statistical power and 0.05 alpha level19. Sixty beta-thalassemia carrier mothers in their gestational age of 11-14 wk with their partners either heterozygous or of unknown thalassemia status comprised the cases. Thalassemia screening was done using high-performance liquid chromatography (HPLC) as a routine procedure for all expectant carrier females with their foetuses at risk of beta-globin gene mutations. CVS from the selected mothers via trans-abdominal chorionic villi sampling (TA-CVS) and blood samples from both parents were collected in the Obstetrics and Gynaecology department at SCB MCH, Cuttack.
Molecular analysis of placental tissue
Extraction of DNA from the CVS was done in the molecular laboratory of the PG department of Biochemistry, SCBMCH, Cuttack as described elsewhere20-22. Briefly, a minimum of 25 mg CVS tissue was washed with normal saline and EDTA and centrifuged at 5000 rpm for 4 min. The supernatant was discarded, leaving the residue. Overnight incubation at 37°C was done in 300 μl lysis buffer and 30 μl proteinase K. Protein precipitating reagent was added and centrifuged at 13000 rpm for 2 min. The supernatant, rich in DNA from placental tissue, was carefully pipetted out to a new tube. The residue containing the precipitated proteins was labelled for estimation of total glutathione (T-GSH), oxidised glutathione (GSSG), and ET-1. Quantity and quality of the DNA and protein in respective samples were assessed using a Nanodrop Spectrophotometer (Thermo Fisher Scientific, USA). Crude DNA extraction from blood samples of both parents was done as per the manufacturer’s instructions (Novaplex™ II β-Thalassemia Assay kit)23.
Genotyping and melting-curve analysis
The β-globin genemutations: IVS 1-1(G>T), 619 bp deletion, IVS 1-5(G>C), CD15(G>A), FS41/42(-TTCT), HbS, FS8/9(+G), -90(C>T), CD16(-C), HbE, CD15(-T), CD30(G>C), and -28(A>G)were characterised using Seegene’s Novaplex™ II β-Thalassemia Assay Kit by TOCE™ technology in a Bio-Rad CFX96 thermocycler23. This involved a rapid temperature increase to 95°C followed by a cooling phase to 55°C lasting for 30 sec, gradual raising of temperature to 85°C at a controlled rate of 0.5°C/s. The melting curve was meticulously analysed during this phase by continuously measuring the fluorescence21,22. Quality control for this analysis included duplicate testing for 20 per cent of samples and the inclusion of positive and negative controls in each assay run. Novaplex™ II β-Thalassemia Assay Kit uses house-keeping genes as internal controls23.
Estimation of T-GSH, GSSG and ET-1
T-GSH, GSSG were estimated by a colorimetric assay (Elabscience kit). Redox status was measured as a ratio of GSH to GSSG13. The placental level of ET-1 was assessed using Elabscience ELISA kits. The principle of these assays involves antigen-antibody interactions, followed by enzymatic reactions that produce a colorimetric signal proportional to the analyte concentration. All assays were performed in triplicates to ensure reproducibility, with positive and negative controls in each run13,24.
Statistical analysis
Chorionic villi samples were divided into wild (without any mutations), heterozygous, and homozygous categories based on the results of β-globin gene mutations. Participant demographics were compiled using Microsoft Excel 2010. The dataset was analysed using SPSS version 29 (IBM; Chicago). Prevalence rates of β-thalassemia cases in homozygous, heterozygous, and wild categories were expressed as percentages. The various genomic mutations of the β-globin gene were delineated in terms of proportions. GSH, GSSG, Redox ratio, and ET1 were represented as median values with interquartile range (IQR). These values were subjected to comparison across homozygous, heterozygous, and wild groups using the Kruskal-Wallis H test, specifically designed for nonparametric datasets. A Pearson correlation (P<0.05) was conducted between GSH and ET1 levels to explore the potential associations between these two variables.
Results
Baseline characteristics, mutation distribution, and redox status among the mutation groups
Baseline characteristics of the study participants and the comparative analysis of GSH, GSSG, redox status, and ET-1 in CVS are shown in table I. Maternal age, Hb, gravida, gestational age, nuchal translucency (NT), and crown-rump length (CRL) were matching among all the groups. Mutation analysis of all 60 cases in the study detected 18 wild, 14 homozygous, and 28 heterozygous foetuses. Maternal Hb levels were lower in mothers with homozygous and heterozygous mutant foetus groups but without significant differences between them. Foetuses with heterozygous HbS and heterozygous IVS1-5(G>C) mutations demonstrated significantly reduced levels of GSH compared to wild or the matched healthy control group (P<0.001). The homozygous HbS group exhibited a significant decrease in the redox ratio compared to the wild type (P=0.006). Non-significant differences were observed in the levels of ET-1 among the different mutation groups (P=0.712) (Table I).
| Beta Thalassaemia mutations (n=60) | Maternal age (yr) |
Gestational Age (wk) |
NT (mm) | CRL (mm) | GSH (μmol/l) (median ± IQR) | GSSG (μmol/l) (median ± IQR) | Redox ratio (median ± IQR) | Endothelin-1 (pg/ml) (median±IQR) |
|---|---|---|---|---|---|---|---|---|
| Wild (n=18) | 27.9±4.9 | 11.72±0.55 | 2.03±0.7 | 54.65±9.8 | 3.52±0.81 | 0.13±0.01 | 27.73±9 | 749.93±95.34 |
| Heterozygous HbS (n=14) | 31.8±3.7 | 12.89±0.87 | 1.98±0.8 | 57.98±6.9 | 1.05±0.34 | 0.11±0.02 | 9.13±2.97 | 841.35±97.98 |
| Heterozygous IVS1-5(G>C) (n=14) | 30.6±2.9 | 11.78±0.65 | 1.97±0.9 | 56.78±6.6 | 1.05±0.34 | 0.12±0.02 | 8.84±5.23 | 881.54±170.82 |
| Homozygous HbS (n=10) | 30.9±3.4 | 13.35±0.23 | 1.97±0.5 | 53.47±7.8 | 1.88±0.34 | 1.88±0.34 | 12.63±1.22 | 902.2±96.32 |
|
Homozygous IVS1-5(G>C) (n=4) |
31.1±2.8 | 12.95±0.28 | 1.96±0.7 | 53.47±7.8 | 1.84±0.29 | 0.12±0.01 | 14.29±2.56 | 858.8±124.99 |
| P value | 0.995 | 1.07 | 1.13 | 0.997 | <0.001* | 0.006* | <0.001* | 0.712 |
P*<0.05 is considered to be significant. Data are mean±standard deviation. n, number of patients; NT, nuchal translucency; CRL, crown-rump length; GSH, glutathione; GSSG, oxidised glutathione; IQR, interquartile range; HbS, sickle haemoglobin
Genotyping of the β-globin gene-associating mutations by melting curve analysis
Melt-curve analysis of HbS homozygous and IVS1-5(G>C) heterozygous mutations have been presented in figure 1A and B, respectively. HbS mutations accounted for 40 per cent (homozygous 17%, heterozygous 23%), while IVS1-5(G>C) mutations constituted 30 per cent (homozygous 7%, heterozygous 23%) of all (Fig. 2A). Analysis of the affected participants revealed that 50 per cent of fathers carried HbS mutation, 41.6 per cent had the IVS1-5(G>C) substitution, and 8.33 per cent presented with the CD15(-T) deletion. In contrast, 53.3 per cent of mothers exhibited the HbS mutation, 40 per cent carried the IVS1-5(G>C) substitution, and 6.7 per cent had the CD15(-T) deletion. Overall, inheritance analysis among all the samples showed 30 per cent of foetuses as wild, and 70 per cent were mutants (Fig. 2B).
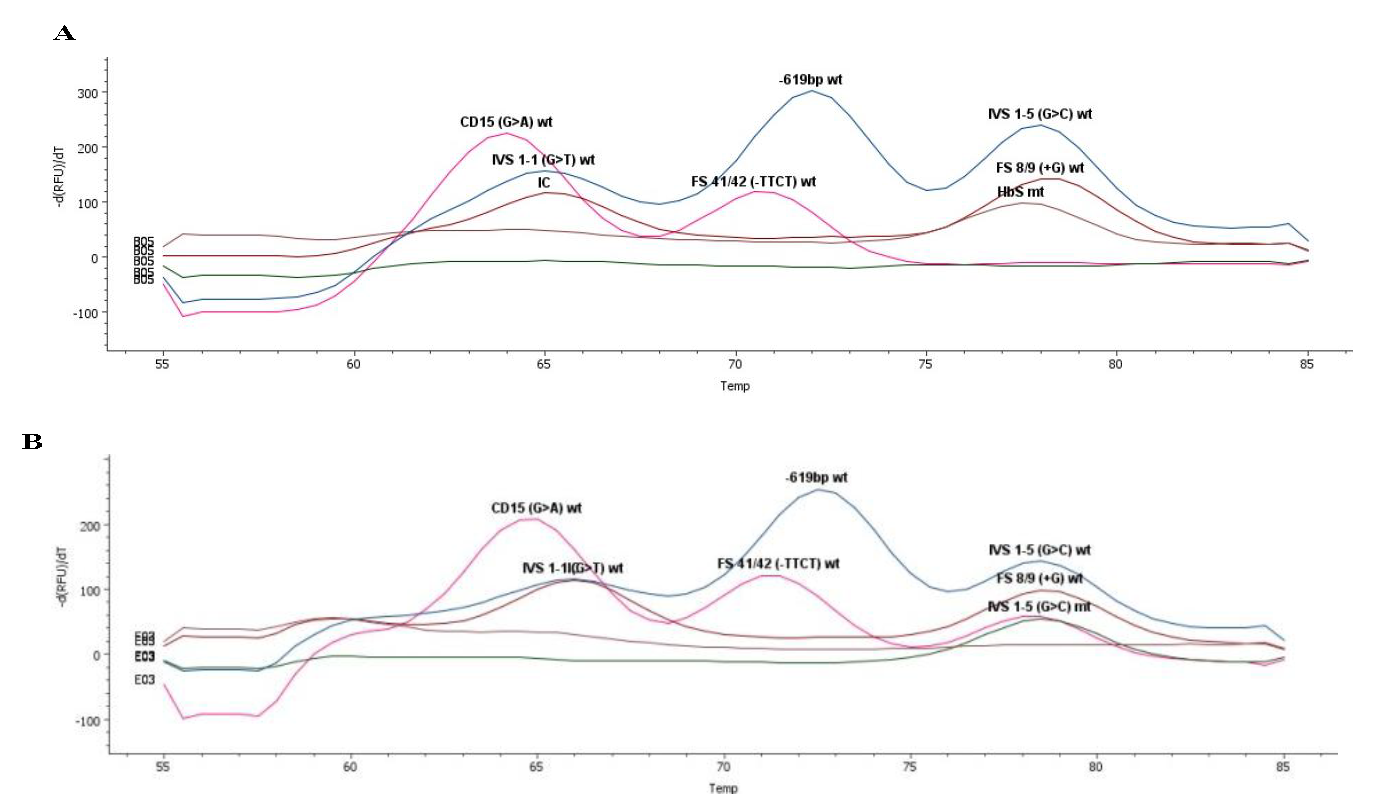
- Melting-curve analysis showing a case of (A) HbS Homo mutation and (B) IVS1-5(G>C) hetero mutation.
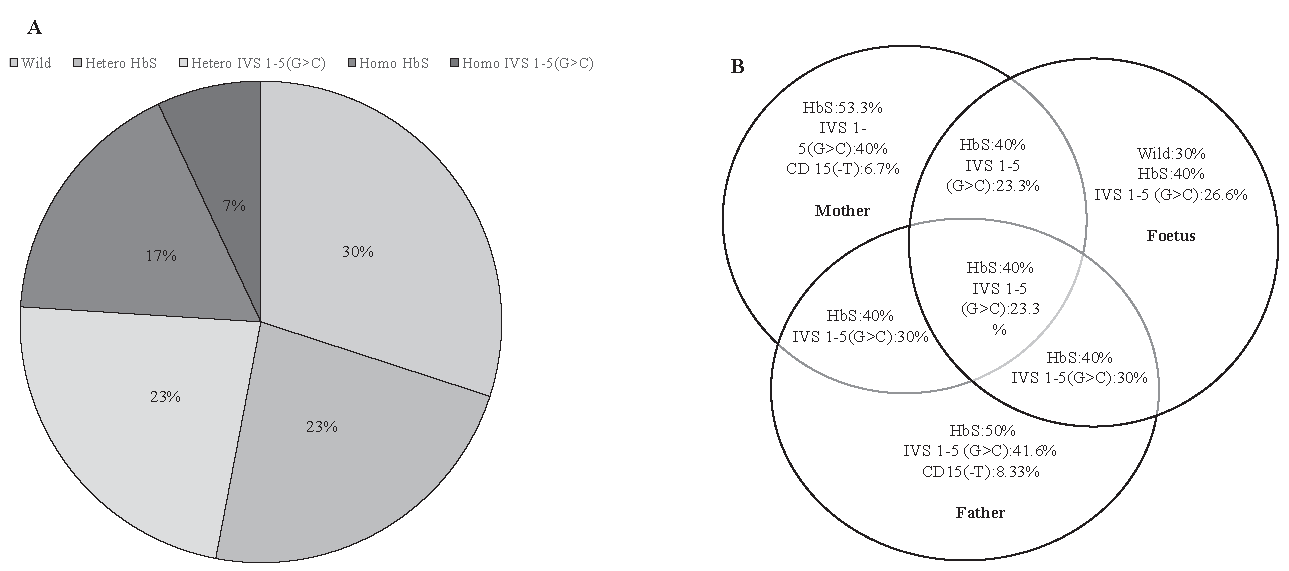
- Distribution of Beta globin gene mutations. (A) Pie chart representing the distribution of Beta globin gene mutations in CVS samples. (B) Distribution of genetic mutations in Beta globin gene mutations among parents and foetuses illustrated by Venn Diagram.
Oxidative stress markers analysis by Kruskal-Wallis test and post hoc analysis
Variations in GSH, GSSG, and redox ratios across the various mutation groups were found to have a non-normal distribution using Shapiro Wilk test. Kruskal Wallis H test was used to compare the medians across the various groups. Assumptions of the Kruskal-Wallis H test, including independent samples and comparable variance distributions, were verified before analysis. The results indicated significant variations in GSH, GSSG, and the Redox ratio (P<0.05) among the mutation groups. The post hoc analysis revealed specific differences among the mutation groups. The wild type group exhibited significantly higher GSH levels compared to heterozygous and homozygous groups (P<0.001). Conversely, the homozygous group had significantly lower GSH levels compared to the wild type (P<0.001) and heterozygous (IVS1-5G>C) (P=0.004) groups (Fig. 3A). GSSG levels were significantly different between the wild type and heterozygous (IVS1-5G>C) groups (P=0.003). The redox ratio was significantly higher in the wild-type group compared to (IVS1-5G>C) heterozygous and homozygous groups (P<0.001). However, the homozygous group exhibited a significantly lower redox ratio compared to the wild (P<0.001) and heterozygous (IVS1-5G>C) (P=0.017) groups (Fig. 3B). ET-1 levels showed non-significant differences among the β-thalassemia mutation groups (P=0.867) (Fig. 3B). A correlation analysis between reduced GSH and ET-1 levels among all subjects presented a correlation coefficient of r=-0.120, P=0.362 (Table II).
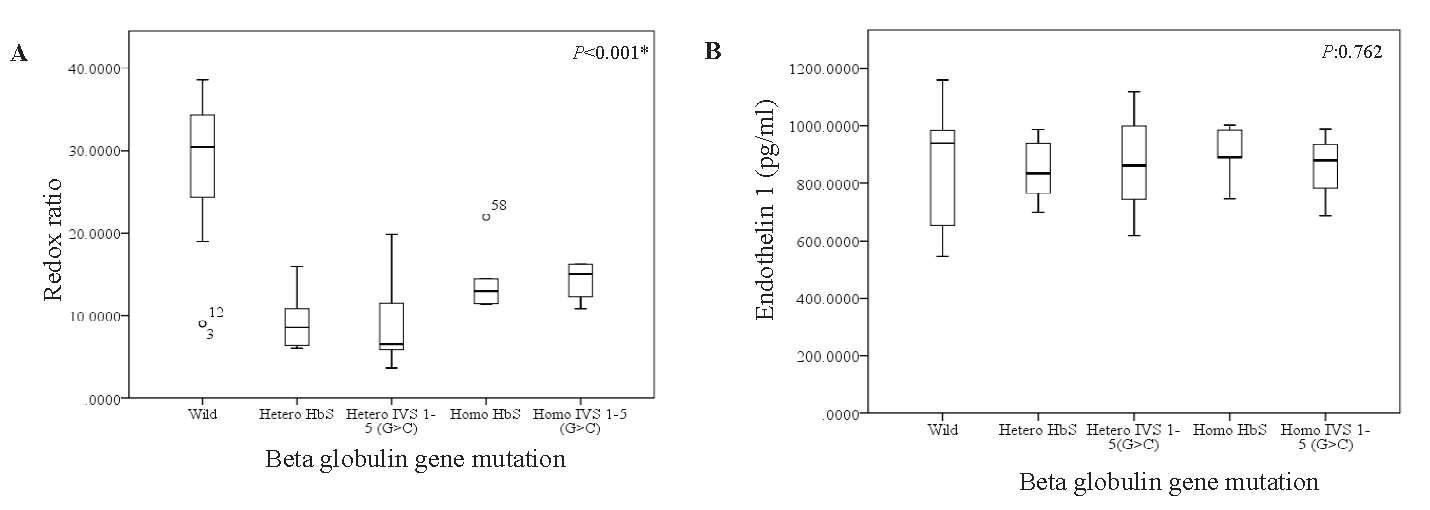
- The Box-whisker plots showing the distribution of the (A) redox ratio and (B) Endothelin-1 across different spectra of Beta Globin gene mutations using SPSS 22.
| Parameters | GSH (μmol/l) | |
|---|---|---|
| r | P value | |
| Endothelin-1 (pg/ml) | -0.120 | 0.362 |
P*<0.05 is considered to be significant
Discussion
Sickle cell disease was ranked as the 12th leading cause of death among children under five yr of age, with an estimated 81,100 deaths25. Pregnancy complications in β-thalassemia carriers, including an increased risk of abortion, small-for-gestational-age (SGA) foetuses, and preterm birth, have been well-documented. A retrospective cohort study found that β-thalassemia carriers had a significantly higher abortion rate, with a relative risk of 3.25 (95% CI: 1.35-7.8) compared to non-carriers26. Additionally, a study assessing pregnancy outcomes in α- and β-thalassemia carriers observed a preterm birth rate of 9.52 per cent in β-thalassemia carriers, suggesting a possible link between the carrier status and preterm delivery27. Further research on pregnancies complicated by β-thalassemia/haemoglobin E disease has demonstrated significant associations with foetal growth restriction, preterm birth, and low birth weight28.
This study investigated the association between specific β-globin gene mutations and oxidative stress biomarkers in β-thalassemia carrier mothers who are at a high risk for having the affected offsprings. The characterisation and screening of mutations inthe study was done by selecting from the previously reported common mutations in India and Odisha9,10.
The unpaired α-globins in erythrocytes in β-thalassemia release free iron, nonheme iron, or hemichrome, catalysing the Fenton reaction and thus triggering excessive ROS production29. These ROS instigate the peroxidation of membrane lipids, proteins, and DNA, inflicting damage at the cellular and organ levels30. Foetal chorionic villus samples in β-thalassemia and sickle hemoglobinopathy foetuses revealed significantly higher oxidative stress levels compared to the wild-type group (Table I). The differences in GSH levels and redox status among various groups corroborate multiple studies. Kalpravidh et al13 and Boudrahem-Addour et al14 noted GSH depletion correlating with disease severity and oxidative stress markers31,32. Morales et al33 further elucidated that the reduction in GSH and the diminished redox ratio in β-thalassemia may be attributed to overconsumption within the antioxidative pathways, contributing to the pathophysiology of the disorder. GSH, empowered by the potent electron-donating capacity of its cysteine residue sulfhydryl groups, serves as a source of reducing equivalents for enzymatic antioxidants like glutathione peroxidase (GPx) and glutathione-S-transferase (GST), undergoing a transition into its oxidised state (GSSG). The dynamic process of converting GSSG back to GSH, orchestrated by glutathione reductase (GR), makes it indispensable for cellular resilience and homeostasis amidst oxidative challenges34,35.
In the present study there were non-significant differences in ET-1 levels among various β-globin gene mutation groups, (P value of 0.867, Table II). This dysregulation aligns with reports by Viprakasit et al35. ET-1 level disparities in β-thalassemia are linked to chronic anaemia-induced tissue hypoxia, prompting compensatory ET-1 release from endothelial cells, known for vasoconstrictive and pro-inflammatory effects. This dysregulation can be further understood in the context of the iron-dependent endothelin convertase enzyme, which plays a crucial role in converting a precursor molecule called big endothelin-1 to form the active peptide, ET-117. Chronic anaemia leads to ET-1 synthesis, as a compensatory response to tissue hypoxia, which, although aiming to regulate vascular tone and maintain blood pressure, may exacerbate vasoconstriction and contribute to vascular complications in conditions like β-thalassemia36. As demonstrated by Patel et al37, placenta growth factor (PlGF) induces ET-1 expression via the activation of hypoxia-inducible factor-1α (HIF-1α), pathways involving reactive oxygen species (ROS) and PI3 kinase. PlGF levels also are intrinsically elevated in conditions associated with chronic anaemia, such as thalassemia and sickle cell disease (SCD), due to increased erythroid turnover37. This is evidenced by elevated ET-1 levels across all mutant groups, with the highest levels observed in homozygous mutations and lower levels in non-mutant foetuses (Table I). Piechota et al17 also uncovered a novel role for ET-1 in preserving erythrocytes. Activation of the ETB receptor by endothelins was found to protect erythrocytes from self-destruction, particularly when triggered by energy depletion. This protective mechanism is achieved by reducing calcium influx and phosphatidylserine exposure17. The lack of differences in ET-1 levels could be a compensatory vascular response or the involvement of alternative oxidative stress pathways. Tantawy’s focus on the influence of oxidative stress on endothelial dysfunction in paediatric β-thalassemia major patients, aligns with our results, revealing a consistent association between GSH and ET-1 levels in β-thalassemia38. Specific intra-group comparisons between heterozygous and homozygous mutations were not explored in detail. Future studies should focus on mutation-specific differences to better understand their impact on vascular health.
Overall, advances in diagnostic precision, early interventions, and preventive measures have transformative implications for foetal outcomes. Novel pharmacological strategies, including small molecule epigenetic modulators, ETB monoclonal antibodies, and the exploration of signalling pathway-biased agonists and antagonists, offer more effective β-management and its associated complications.
Acknowledgment
Authors express their sincere gratitude to the participants who generously contributed to this research.
Financial support & sponsorship
None.
Conflicts of Interest
None.
Use of Artificial Intelligence (AI)-Assisted Technology for manuscript preparation
The authors confirm that there was no use of AI-assisted technology for assisting in the writing of the manuscript and no images were manipulated using AI.
References
- Draft policy for prevention and control of hemoglobinopathies – thalassemia, sickle cell disease, and variant hemoglobins in India. New Delhi, 2018. Available from: https://mohfw.gov.in/sites/default/files/drft%20policy.pdf, accessed on March 16, 2025.
- Spectrum of hemoglobinopathies: A new revelation in a tertiary care hospital of Odisha. Indian J Hematol Blood Transfus. 2019;35:513-7.
- [Google Scholar]
- Spectrum of hemoglobin disorders in Southern Odisha, India: A hospitalbased study. Porto Biomed J. 2021;6:e126.
- [Google Scholar]
- Global epidemiology of hemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86:417-96.
- [Google Scholar]
- Prevention and control of haemoglobinopathies in India-Thalassaemia, sickle cell disease and other variants haemoglobin. 2016. Available from: https://nhm.gov.in/images/pdf/programmes/RBSK/Resource_Documents/Guidelines_on_Hemoglobinopathies_in%20India.pdf, accessed on March 16, 2025.
- IthaGenes: An interactive database for haemoglobin variations and epidemiology. PLoS One. 2014;9:e103020.
- [Google Scholar]
- Molecular genetics of β-thalassemia: A narrative review. Medicine (Baltimore). 2021;100:e27522.
- [Google Scholar]
- Profile of beta-thalassemia in eastern India and its prenatal diagnosis. Prenat Diagn. 2004;24:992-6.
- [Google Scholar]
- Molecular variants and clinical importance of β-thalassaemia traits found in the State of Orissa, India. Hematology Am Soc Hematol Educ Program. 2009;14:290-6.
- [Google Scholar]
- Metabolomic investigation of β-thalassemia in chorionic villi samples. J Clin Med. 2019;8:798.
- [Google Scholar]
- Oxidative stress in placenta: Health and diseases. Biomed Res Int. 2015;2015:293271.
- [Google Scholar]
- Glutathione system in animal model of solid tumors: From regulation to therapeutic target. Crit Rev Oncol Hematol. 2018;128:43-57.
- [Google Scholar]
- Oxidative stress in β-thalassaemia and sickle cell disease. Redox Biol. 2015;6:226-39.
- [Google Scholar]
- Significance of polymorphisms and expression of enzyme-encoding genes related to glutathione in hematopoietic cancers and solid tumors. Biomed Res Int. 2015;2015:853573.
- [Google Scholar]
- Activation of multiple signal transduction pathways by endothelin in cultured human vascular smooth muscle cells. Eur J Biochem. 1990;189:415-21.
- [Google Scholar]
- Role of endothelin-1 receptor blockers on hemodynamic parameters and oxidative stress. Pharmacol Rep. 2010;62:28-34.
- [Google Scholar]
- G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175-91.
- [Google Scholar]
- DNA extraction from chorionic villi for prenatal diagnosis of foetal haemoglobin genotype. West Afr J Med. 2011;30:400-3.
- [Google Scholar]
- Dual priming oligonucleotide system for the multiplex detection of respiratory viruses and SNP genotyping of CYP2C19 gene. Nucleic Acids Res. 2007;35:e40.
- [Google Scholar]
- TOCE™ high multiplex real-time PCR. Available from: https://www.seegene.com/technologies/toce, accessed on July 24, 2024.
- Enzyme immunoassay and enzyme-linked immunosorbent assay. J Invest Dermatol. 2013;133:e12.
- [Google Scholar]
- Global, regional, and national prevalence, and mortality burden of sickle cell disease, 2000-2021: a systematic analysis from the global burden of disease study 2021. Lancet Haematol. 2023;10:e585-99.
- [Google Scholar]
- Pregnancy outcomes among women with beta-thalassemia trait. Arch Gynecol Obstet. 2016;293:771-4.
- [Google Scholar]
- The prevalence and outcomes of α- and β-thalassemia among pregnant women in Hubei province, central China: An observational study. Medicine (Baltimore). 2022;101:e28790.
- [Google Scholar]
- Impacts of β-thalassemia/hemoglobin E disease on pregnancy outcomes. Int J Gynaecol Obstet. 2024;166:360-7.
- [Google Scholar]
- Thalassemic erythrocytes release microparticles loaded with hemichromes by redox activation of p72Syk kinase. Haematologica. 2014;99:570-8.
- [Google Scholar]
- Oxidative stress and β-thalassemic erythroid cells behind the molecular defect. Oxid Med Cell Longev. 2013;2013:985210.
- [Google Scholar]
- Glutathione redox system in β-thalassemia/Hb E patients. Scientific World Journal. 2013;2013:543973.
- [Google Scholar]
- Oxidative status and plasma lipid profile in β-thalassemia patients. Hemoglobin. 2015;39:36-41.
- [Google Scholar]
- Iron chelation therapy with deferiprone improves oxidative status and red blood cell quality and reduces redox-active iron in β-thalassemia/hemoglobin E patients. Biomed Pharmacother. 2022;145:112381.
- [Google Scholar]
- Crosstalk between cytokine profile, redox, and iron status in β-thalassemia: Relation to frequency/duration of blood transfusion. Pediatr Hematol Oncol. 2019;36:151-60.
- [Google Scholar]
- Baseline levels of plasma endothelin-1 (ET-1) and changes during transfusion in thalassemic patients. Am J Hematol. 2002;70:260-2.
- [Google Scholar]
- The role of endothelin-1 and endothelin receptor antagonists in the inflammatory response and sepsis. Arch Immunol Ther Exp (Warsz). 2015;63:41-52.
- [Google Scholar]
- Placenta growth factor augments endothelin-1 and endothelin-B receptor expression via hypoxia-inducible factor-1α. Blood. 2008;112:856-65.
- [Google Scholar]
- Endothelin-1 gene polymorphism (G8002A) and endothelial monocyte-activating polypeptide II: Role in vascular dysfunction in pediatric patients with β-thalassemia major. Cytokine. 2023;161:156048.
- [Google Scholar]




