Translate this page into:
Aetiologic spectrum of mental retardation & developmental delay in India
Reprint requests: Dr Shubha R. Phadke, Professor, Department of Medical Genetics, Sanjay Gandhi Post Graduate Institute of Medical Sciences, Lucknow 226 014, India e-mail: shubharaophadke@gmail.com
-
Received: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
The aetiology of mental retardation is varied and difficult to establish. Reports from India on the spectrum of underlying causative conditions are lacking. This retrospective study was conducted to establish the various aetiologies of mental retardation (MR) and developmental delay (DD) in patients attending a medical genetics centre in north India and to assess the contribution of genetic disorders.
Methods:
This retrospective study was conducted at a tertiary care centre in north India. All patients attending the centre with MR or DD from January 2007 to December 2009 were included. The aetiology of MR/DD was ascertained after clinical assessment and targeted laboratory evaluation. The spectrum of causative conditions and contribution of genetic disorders was established.
Results:
A total of 338 patients were included in the study, of whom definite diagnosis was established in 253 (74.8%). The various aetiological categories were: chromosomal disorders in 112 (33.1%), non chromosomal syndromes in 32 (9.5%), neurometabolic disorders in 34 (10.1%), central nervous system structural defects in 25 (7.4%), cerebral palsy in 43 (12.7%) and environmental insults in 7 (2%). Eighty five patients (25.2%) had idiopathic mental retardation. A total of 196 (58%) patients had a genetic disorder as the cause of MR/DD.
Interpretation & conclusions:
The aetiology of MR/DD is varied and difficult to establish in a significant proportion of patients. Chromosomal and various monogenic disorders contribute to a large number of MR/DD cases and hence a genetic work up is essential for all such patients.
Keywords
Aetiology
developmental delay
genetic diseases
India
mental retardation

Mental retardation (MR) occurs in 1-3 per cent of the population12. As per WHO, MR is defined as intellectual function significantly lower than average, with an intelligence quotient (IQ) equal to or less than 70; and poor adaptive skills in at least two of the following areas: communication, self care, social/interpersonal skills, self-guidance, school performance, work, leisure, health and safety; and onset of symptoms before the age of 18 yr3. An IQ greater than 85 is considered normal, and individuals with an IQ between 71 and 84 are regarded as having a borderline IQ level4. Individuals with an IQ of 50 to 70 have mild MR; those with an IQ of 35 to 50, moderate MR; those with an IQ of 20 to 35, severe MR; and those with an IQ lower than 20, profound MR4. Mild MR is seven to ten times more common than moderate or severe MR4. In children below 5 yr age, formal IQ assessment is not possible and the term developmental delay (DD) is used to identify children with a subset of developmental disabilities defined as significant delay in two or more of the following developmental domains: gross/fine motor, speech/language, cognition, social/personal, and activities of daily living5. Though both these terms (MR and DD) are used synonymously, not every child with developmental delay has mental retardation when tested formally at a later age5.
Varying aetiologies contribute to MR/DD, and their accurate identification is essential for prognostication and management of the patient and for providing recurrence risk estimates and prenatal diagnosis options to the families36. Standard guidelines for the evaluation of an individual with MR/DD have been provided by American College of Medical Genetics6, American Academy of Neurology and The Practice Committee of the Child Neurology Society5 and American Academy of Pediatrics7. The diagnostic yield of these guidelines has been reported to range from 40 to 60 per cent. It is usually higher in patients with severe MR as compared to mild MR5–9.
In the present study, the patients with MR/DD attending a tertiary care genetic centre in north India were evaluated. It was also attempted to classify them into diagnostic categories and estimate the contribution of genetic diseases to MR/DD. A targeted investigation protocol was followed keeping in mind resource constraints and to provide a model for investigation of such patients in a developing nation.
Material & Methods
This retrospective study was conducted at the Department of Medical Genetics, Sanjay Gandhi Post Graduate Institute of Medical Sciences (SGPGIMS), a tertiary care centre at Lucknow, the capital of Uttar Pradesh. The patients attending the outpatient clinic of the department with complaints of MR/DD from January 2007 to December 2009 were included in the study. The department is a referral centre for genetics services and the patients hailed from various parts of India, although most of them belonged to the surrounding States. All patients were examined by a clinical geneticist with detailed history, dysmorphology evaluation and neurological examination. Development assessment was done for children <3 yr by Developmental Assessment Scale for Indian Infants (DASII)10. For older children, IQ assessment was done using Binet-Kamat test of intelligence (2-18 yr) and Malin's Intelligence Scale for Indian Children (MISIC, 6-15 yr)11.
Availability of diagnostic tests and financial constraints were main considerations taken into account to design the diagnostic algorithm. There was no single panel of tests done for all the patients, rather the clinical findings were used to narrow the possible diagnoses, which were then investigated. Investigative techniques like microarray were not available for patient care at our centre. The karyotype was done at 550 band resolution. Neuroimaging techniques included CT scan and/or MRI. Fluorescence in situ hybridization (FISH) testing was done using specific probes according to the phenotype. Metabolic testing included appropriate enzyme studies and gas chromatography mass spectrometry (GCMS) and Tandem mass spectrometry (MSMS) where indicated. Urine spot test was a screening test for phenylketonuria, galactosemia, homocystinuria and mucopolysaachridoses.
On the basis of clinical and investigation findings diagnosis of the patients was established. They were grouped according to underlying pathology into categories of (i) Chromosomal disorders: Numerical as well as structural rearrangements including microdeletion syndromes; (ii) Non chromosomal syndromes; (iii) Neurometabolic disorders; (iv) Structural central nervous system defects; (v) Static encephalopathy suggestive of cerebral palsy; (vi) Environmental insult; (vii) Idiopathic mental retardation. The proportion of cases in each category was established. An analysis of the genetic and non-genetic basis of these diagnostic classes was performed.
Results
A total of 338 patients (228 males and 110 females; male : female ratio 2.1 : 1) with mental retardation/developmental delay were included in the study. The average age of patients was 4.75 yr. Excluding the Down syndrome patients (n=97), many of whom were referred at ages as early as one month, the average age of the patients was 5.23 yr ranging from 4 months to 28 yr. IQ assessment was available for 194 of 241 non-Down syndrome patients. The average IQ was 39.8. Seventy four (38.4%) patients had mild MR, 33 (17%) had moderate MR, 71 (36.4%) had severe and 15 (7.7%) had profound MR. One patient had borderline MR. The broad diagnostic categories to which the patients were divided following workup are shown in Table I.
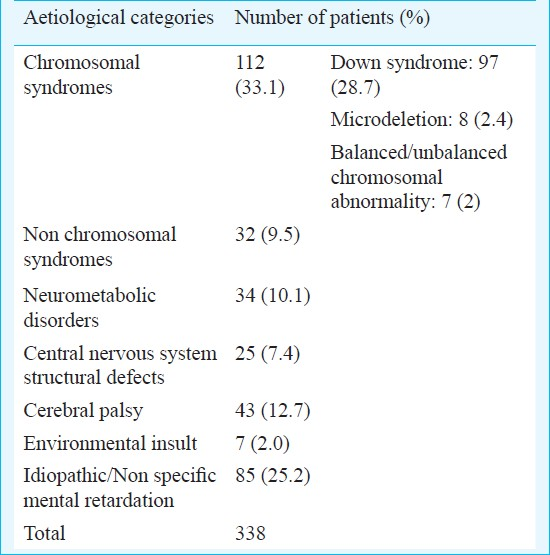
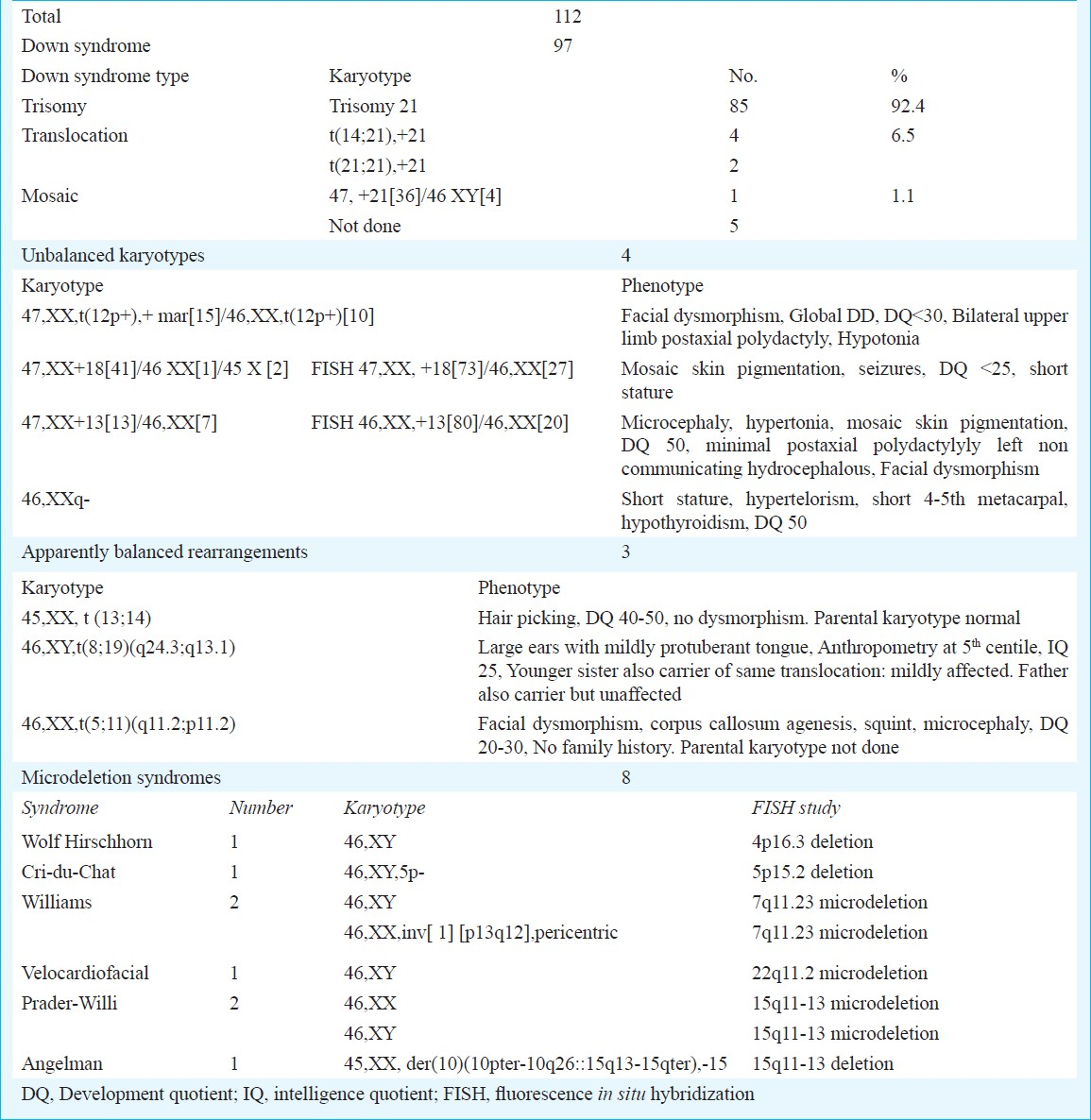
Down syndrome present in 97 (28.7%) patients, comprised the single most common aetiology of MR/DD. Karyotype was done in 92 patients with Down syndrome, of whom 85 (92.5%) had trisomy 21, four had t(14;21), two had t(21;21) (translocation Down syndrome in 6.5%) and one (1.1%) was mosaic for trisomic and disomic cell lines. Other chromosomal abnormalities consisting of unbalanced karyotypes (4), apparently balanced chromosomal rearrangements (3) and microdeletion syndromes (8) together accounted for 15 (4.5%) patients. The karyotypes and FISH results of these patients are depicted in Table II. The patients with microdeletion syndromes had phenotypes characteristic of the respective syndrome. Except two, all patients with unbalanced karyotypes had a de novo abnormality. One patient with Angelman syndrome had a mother with balanced translocation involving the 15q11.2 region. The other patient, a boy had apparently balanced translocation t (8; 19) which was also present in his mentally retarded sister and they had inherited it from their father who was phenotypically normal. In patients with cytogenetically balanced chromosomal rearrangement no other cause of mental retardation was detected.
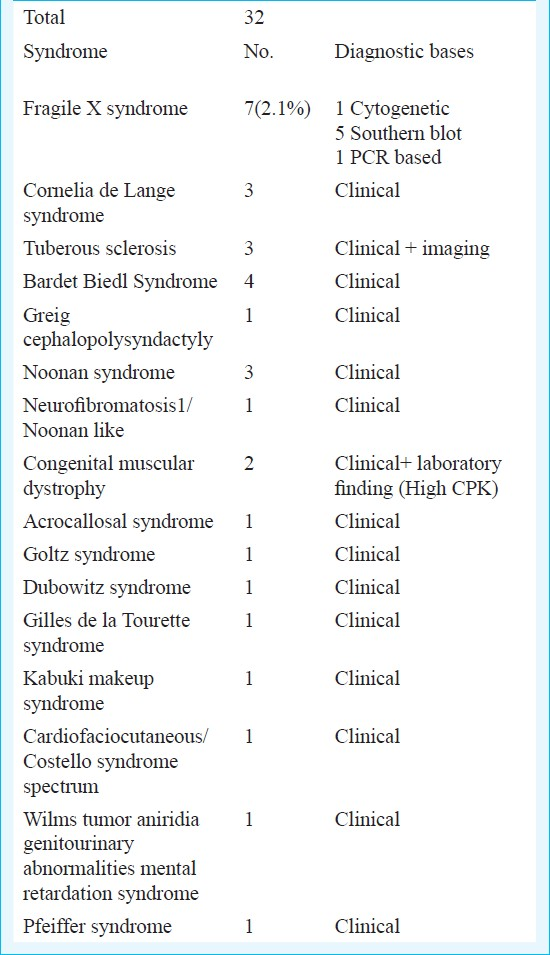
Thirty two patients had various monogenic or sporadic non chromosomal syndromes (Table III). The majority of syndromes were diagnosed clinically. Although molecular confirmation was available for many of these disorders, cost constraints prevented us from availing this. Fragile X syndrome was found in 7 (2.1%) patients and confirmed by Southern blot in most.
Thirty four patients had neuroregression or progressive neurological disease suggestive of a neurometabolic disorder (Table IV). Mucopolysaachridosis was found in 6, sphingolipidosis in 6 and leukodystrophy in 10 patients. One patient each had late infantile neuronal ceroid lipofuscinosis, two hydroxy glutaric aciduria and aromatic L amino acid decarboxylase deficiency. In eight patients clinical diagnosis could not be confirmed due to various reasons. One patient had Maroteaux-Lamy syndrome, which is not reported to be associated with MR.

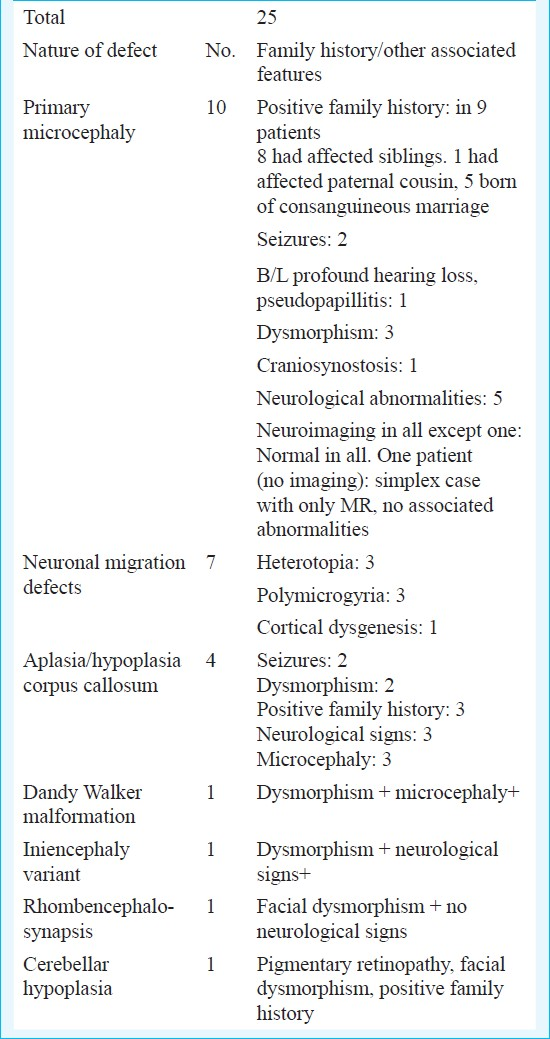
Structural abnormalities of the central nervous system were found in 25 (7.5%) patients (Table V). Microcephaly vera, which was defined as occipitofrontal diameter less than 3 SD for age and sex with no structural malformation of brain, was found in 10 patients. Nine of these had positive family history, 8 having similarly affected sibling and one with affected paternal cousin. Five patients had associated neurological complications like motor deficits, seizures and ophthalmic or auditory complications (present in 1 patient). Seven patients had neuronal migration defects and 4 had agenesis of corpus callosum, three of whom had positive family history and two had associated dysmorphism.
Seven patients had acquired causes in the form of psychosocial deprivation and TORCH group of infection as the causative condition.
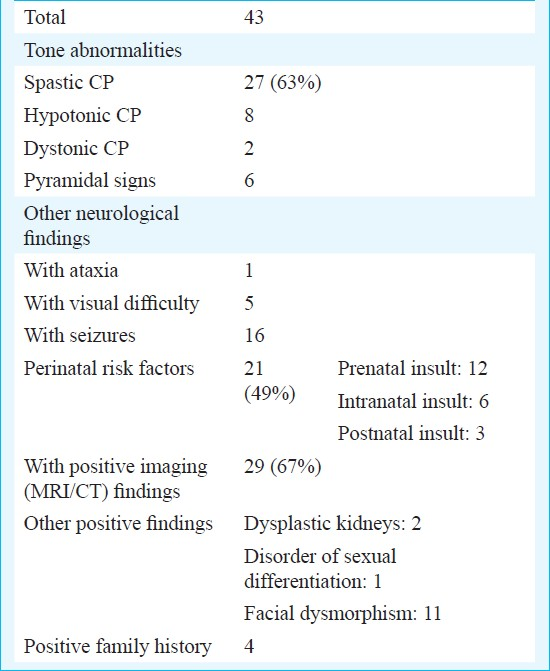
Forty three (12.7%) patients had static encephalopathy with motor deficit and were diagnosed as cerebral palsy. In 29 of these there was evidence of hypoxic brain injury on neuroimaging whereas for the remaining 14 the diagnosis was based on the history and examination findings. In 21 patients history of perinatal insult or risk factors was present (Table VI).
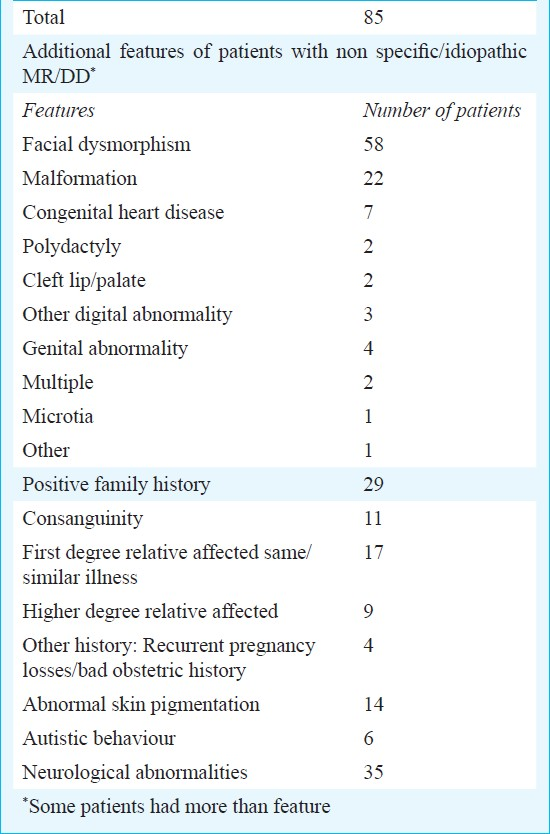
For 85 (25.1%) patients, no definite diagnosis could be established, 58 had dysmorphic features, 22 had various malformations, 29 had positive family history and 35 had neurological findings (Table VII).
Discussion
In the present study 338 patients with MR/DD were assessed. Males were two times more common, a finding similar to but slightly higher than previous literature reports13. This high proportion of males can be due to the fact that female children are less likely to be brought for evaluation of MR due to various social and cultural factors. The prevalence of severe MR was as high as 36 per cent and and almost similar to mild MR. This is contrary to expectations, but can be explained by an ascertainment bias, whereby patients with more severe disability are more likely to seek medical attention and more likely to be referred to a geneticist. Also, patients with mild MR are likely to be labelled as just poor learners and not really brought to a doctor for the fear of stigmatisation.
The contribution of different aetiologies to MR/DD was similar to previous literature reports. Chromosomal disorders constituted the largest group of patients (33.1%), reported literature indicated 15-40 per cent cases of MR/DD attributed to chromosomal disorders16712–16. Down syndrome was the most common single diagnosis, occurring in one third of patients, a figure also in corroboration with previous studies1314. About 2 per cent patients were found with other chromosomal abnormalities and 2.4 per cent patients with microdeletion syndromes detected by FISH studies. Previous studies have revealed cytogenetically visible chromosomal abnormalities other than trisomy 21 in 1.1-11 per cent patients713–17. Microdeletion syndromes have been reported in 1-19 per cent of MR cases in different studies681516. The three patients with apparently balanced chromosomal rearrangements, may have small genomic imbalances at the chromosomal breakpoints or may have other unrelated cause for MR.
The non chromosomal genetic syndromes consisted of a variety of heterogenous single gene disorders. The largest number was of Fragile X syndrome found in 2.1 per cent patients, similar to previously reported prevalence of 1-5 per cent57812. Fragile X testing may be guided by the clinical picture. One study from India found Fragile X syndrome in 6.3 per cent of boys with MR and the authors felt that clinical scoring could help in decision making regarding Fragile X testing18. For most other syndromes the diagnosis was clinical. Many common genetic conditions like Tuberous sclerosis, Cornelia de Lange syndrome, Bardet Biedl syndrome, Noonan syndrome and congenital muscular dystrophy were found in more than one patient. Overall, this group comprised of 9.5 per cent of all patients with MR/DD. Previous reports indicate figures of 6-21 per cent26131517.
Neurometabolic disorders were present in around 10 per cent patients in this study, which was higher as compared to previous reports. Most studies conclude that neurometabolic disorders constitute 1-5 per cent of MR/DD cases26121315–17 although Shevell et al5 indicated a yield of 14 per cent by targeted metabolic studies. In the present study, leukodystrophies accounted for upto one third of these cases, metachromatic leukodystrophy being the commonest subtype. Mucopolysaachridosis and GM1 and GM2 gangliosidosis were present in majority of remaining cases. Large proportion of consanguineous marriages in the Indian subcontinent may be the reason for high prevalence of these primarily autosomal recessive disorders.
CNS anomalies were diagnosed in 10 per cent patients, similar to previous studies where diagnostic abnormalities on neuroimaging were found in 2-40 per cent patients with MR1567121417. Microcephaly vera was diagnosed in 10 patients, of whom half had associated neurological abnormalities. Although majority of microcephaly vera patients do not have neurological abnormalities, the high proportion of these in our study may be due to a referral and ascertainment bias. Corpus callosum hypoplasia and other rare malformations may be of heterogeneous aetiology, but the presence of dysmorphism and/or positive family history in most of these patients indicated predominant genetic contribution.
A significant proportion of patient had static encephalopathy with motor deficit, indicating a diagnosis of cerebral palsy. Cerebral palsy has been reported to contribute 2.5 to 23 per cent of mental retardation cases in different groups of MR patients215–17. Majority of these (63%) had spastic cerebral palsy, as has been reported earlier19. Previous studies have also concluded prenatal insult as the commonest cause of cerebral palsy20. Intranatal events suggestive of birth asphyxia are believed to contribute to 8-15 per cent of cases20. In our study 28 per cent cases were found with history of an adverse intranatal event. Some patients in our study also had facial dysmorphism and/or positive family history suggesting possible genetic predisposition to their illness. We had a large proportion of cases (67%) in our study where evidence of CNS insult on neuroimaging could be found.
Acquired causes like prenatal TORCH group of infection and psychosocial deprivation were also found in a few patients in this study. One patient had possible intracranial hypoxia following hypocalcemic seizures induced by rickets. Different studies and reviews in the past have indicated a wide range of contribution from various environmental factors ranging from 1-25 percent1615–17.
A definite diagnosis of MR/DD may not be possible in 30-60 per cent patients, although figures ranging from 10-80 per cent have been reported in different studies. Finding a definite cause is especially difficult in mild MR cases where psychosocial factors play a major role125–913–17. In the present study, no diagnosis could be established in one fourth of the patients, indicating that in at least 75 per cent of patients a definite cause of MR/DD was found. We had several cases of severe MR, which may explain the high diagnostic yield in our setting. Also, targeted evaluation based on the clinical findings helped in maximising the diagnostic yield. Further investigations like multiplex ligation probe assay (MLPA) for common microdeletion syndromes, for subtelomeric regions and for X linked MR loci, MECP2 gene study and array based cytogenetic analysis need to be done to identify the aetiology in the large group of patients with unexplained MR/DD. Various clinical clues like the presence of dysmorphism, malformation, autism spectrum disorder and positive family history indicate high probability of finding a genetic aetiology with these new techniques.
Overall, 58 per cent patients in this study had a definite genetic disorder contributing to MR/DD. For many other patients also, like central nervous system malformations and patients with unexplained MR with malformations/dysmorphism or positive family history, a genetic aetiology is most likely. Previous studies have indicated that genetic disorders contribute to 17-47 per cent of MR7131516.
There is a paucity of reports from India on the spectrum of mentally retarded patients. The only study in recent times is 20 years old, does not delve into clinical details and excludes many categories13. The paucity of diagnostic services has been a major deterrent to the availability of comprehensive reports from this country.
To conclude, we have reported the etiologic spectrum of patients with MR/DD at our centre. We have found a large proportion of severe MR cases was found with a significant contribution of genetic disorders to the clinical presentation. This indicates the need to establish genetic services as part of paediatric care or integration of such services as part of care in institutions for mentally challenged individuals. The diagnosis of these genetic disorders is important for management of these persons as well as offering prenatal diagnosis services to the family. A high yield of a targeted diagnostic approach was also observed in the workup of these patients. Hence, in the setting of financial and resource constraints, the availability of appropriate investigations as well individuals with expertise to judiciously utilize such investigations are essential to evaluate patients with MR/DD.
Acknowledgment
Authors thank the Indian Council of Medical Research, New Delhi, for financial support in providing diagnostic services to our patients and also acknowledge the help provided by Dr Thelma, South Campus, University of Delhi, New Delhi, for providing Southern blot testing for Fragile X syndrome.
References
- Mental retardation: definition, classification and systems of supports. Washington DC: AAMR; 1992.
- Diagnostic evaluation of developmental delay/mental retardation: An overview. Am J Med Genet C Semin Med Genet. 2003;117C:3-14.
- [Google Scholar]
- Practice parameter: evaluation of the child with global developmental delay. Neurology. 2003;60:367-80.
- [Google Scholar]
- Evaluation of mental retardation: recommendations of a Consensus Conference: American College of Medical Genetics. Am J Med Genet. 1997;72:468-77.
- [Google Scholar]
- and the Committee on Genetics. Clinical genetic evaluation of the child with mental retardation or developmental delays. Pediatrics. 2006;117:2304-16.
- [Google Scholar]
- American College of Medical Genetics guideline on cytogenetic evaluation of the individual with developmental delay and mental retardation. Genet Med. 2005;7:650-4.
- [Google Scholar]
- Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet. 2006;140:2063-74.
- [Google Scholar]
- Mental and motor growth of Indian babies (1-30 months), Final report. Baroda: Department of Child Development, MSUB; 1970. p. :1-185.
- Malin's intelligence scale for Indian children (MISIC) Indian J Mental Retard. 1971;4:15-25.
- [Google Scholar]
- Diagnostic investigations in individuals with mental retardation: a systematic literature review of their usefulness. Euro J Hum Genet. 2005;13:6-25.
- [Google Scholar]
- ICMR Collaborating Centres & Central Co-ordinating Unit. Multicentric study on genetic causes of mental retardation in India. Indian J Med Res. 1991;94:161-9.
- [Google Scholar]
- A genetic diagnostic survey in a population of 202 mentally retarded institutionalised patients in the south of Brazil. Clin Genet. 1998;54:219-23.
- [Google Scholar]
- Diagnostic yield of the comprehensive assessment of developmental delay/mental retardation in an institute of child neuropsychiatry. Am J Med Genet. 1999;82:60-6.
- [Google Scholar]
- An epidemiological and aetiological study of children with intellectual disability in Taiwan. J Intellectual Disability Res. 1998;42:137-43.
- [Google Scholar]
- Etiologic evaluation in 247 children with global developmental delay at Istanbul, Turkey. J Trop Pediatr. 2005;51:310-3.
- [Google Scholar]
- Prevalence of Fragile-X (A) syndrome in mentally retarded children at a genetics referral centre in Delhi, India. Indian J Med Res. 1998;108:12-6.
- [Google Scholar]
- Clinical spectrum of cerebral palsy in north India - An analysis of 1000 cases. J Trop Pediatr. 2002;48:162-6.
- [Google Scholar]




