
Translate this page into:
Additional markers for genetic diagnosis of type 3 von Willebrand disease in Indian population
*For correspondence: shrimatishetty@yahoo.com
This is an open access article distributed under the terms of the Creative Commons Attribution NonCommercial ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Sir,
von Willebrand disease (VWD) encompasses a wide spectrum of bleeding disorders with severity ranging from moderate bleeding tendency to severe life threatening haemorrhage, estimated to affect approximately 0.5 - 2 per cent of the population in the Western countries1. Patients present with mucocutaneous bleeding symptoms, epistaxis, mucosal bleeding, prolonged bleeding from cuts, post-dental extraction bleeding, menorrhagia and gastrointestinal bleeding. These are classified as types 1, 2 and 3, depending on the qualitative and quantitative defects in von Willebrand factor (VWF) antigen. Diagnosis, genetic counselling, carrier and antenatal diagnosis play an important role in the comprehensive management of these cases.
Identification of mutations in von Willebrand factor gene (VWF) is hampered by the large size and the highly polymorphic nature of the gene, heterogeneity of mutations and a pseudogene corresponding to a large part of the gene2. The spectrum of VWF mutations that cause VWD includes frame shift mutations resulting from small insertions or deletions, large deletions, missense mutations affecting single amino acid residues, splice-site mutations, duplications, nonsense mutations causing premature termination of translation and gene conversion due to recombination between the gene and pseudogene3. Even after sequencing the entire coding region, splicing region and the promoter regions of VWF, in a few cases the mutations are undetected45. It has been reported that analysis of the essential regions of genomic DNA may not be adequate to identify the underlying molecular pathology in a proportion of type 3 VWD cases. Deep intronic mutations affecting mRNA splicing by creation of a cryptic splice site or introduction of additional in-frame exons are some of the plausible explanations for the disease manifestation, which may be detected by mRNA analysis in at least a few cases6. Cost of DNA sequencing to screen 52 exons remains a major obstacle in implementing direct sequencing in the routine genetic diagnosis of VWD.
Restriction fragment length polymorphism (RFLP) technique has been the method of choice for offering carrier detection and antenatal diagnosis in VWD families, particularly in developing countries like India78. The limitations of the technique are the requirement of all the family members and non-informativeness or homozygosity of the polymorphic markers. Heterozygosity of the polymorphisms varies in different populations and it is essential that this is analyzed prior to its application in the genetic diagnosis of the families.
A large number of RFLP markers have been identified for tracking the affected allele in VWF910111213141516. The most useful markers are the highly polymorphic tetranucleotide repeats i.e. variable number of tandem repeats (VNTR) of ATCT repeats in intron 40 of the VWF. These have been successfully used in prenatal diagnosis of VWD by different workers with different rates of heterozygosity. However, in a gene which is as complex as VWF, analysis of heterozygosity frequency of additional markers is always an added advantage to make the genetic diagnosis applicable to majority of the families.
In the present study, we analyzed the entire VWF coding region including intron-exon boundaries by DNA sequencing in 100 unrelated severe VWD patients. All consecutive severe type 3 VWD cases (VWF:Ag < 5 IU/dl and FVIII levels < 10 IU/dl)17 were included in the study. These patients attended the Comprehensive Haemophilia Care Center at the National Institute of Immunohaematology, Mumbai, India, between 2009 and 2013. All cases with acquired von Willebrand syndrome were excluded from the study. The study was approved by the Institutional Ethics committee.
Genomic DNA was extracted using commercial kits (Invitrogen, USA). Fifty five sets of primers for 52 exons were designed in-house, using UCSC (http://genome.ucsc.edu), primer 3 output and the sequence from Ensembl18. The primers were obtained from Sigma Aldrich, USA. Primers were designed so as to include the entire exonic areas, promoter region, as well as exon–intron boundaries and minimum 50-100 bases upstream or downstream to exonic region. Oligo analysis was done for each set of primer to avoid looping or dimer formation. The primers for known Arginine hot spot region were adapted19. To amplify the specific gene without co-amplification of pseudogene, the primers were designed in such a way that those were specific to authentic VWF gene by 1-3 nucleotide mismatches at the region specific to gene and the PCR conditions were optimized for each exon to give authentic gene specific amplification. The DNA sequencing was performed in ABI Prism 3130 Genetic Analyzer (ABI, USA).
The nucleotide and amino acid numbering of each polymorphism was determined by reference to the published VWF sequence20. Sequence variants have been checked manually by identifying the base position on the cDNA sequence. Previously reported single nucleotide polymorphisms were checked in the Ensembl database18.
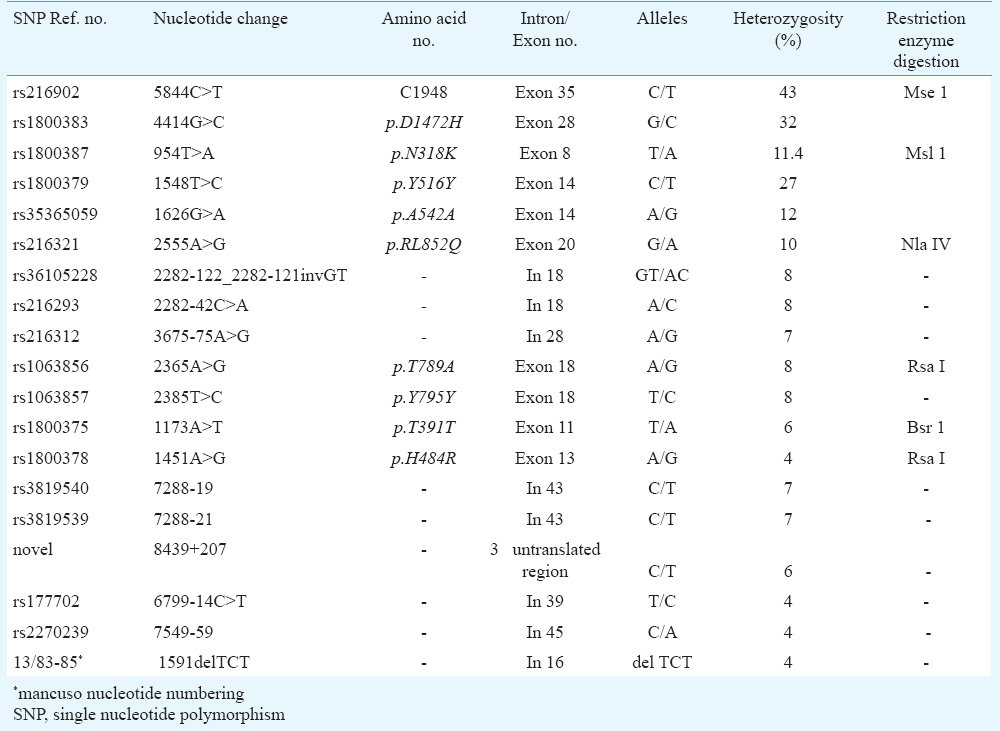
Several intronic and exonic single nucleotide variations (SNVs) were found with different rates of heterozygosity (Table). Most of the variations were intronic and a few silent synonymous changes in exonic region. The c.5844C>T SNV (rs216902), i.e. in the splice site of exon 35 was found to be heterozygous in 43 per cent of our patients. The heterozygosity rates of polymorphic markers p.D1472H, p.N318K, p.T789A, p.R852R, rs177702 C/T, rs216312 A/G, p.T391T and p.H484R polymorphisms were found to be 32, 11.4, 8, 10, 4, 7, 6 and 4 per cent, respectively. The two polymorphisms i.e. p.T789A (rs1063856) and p.Y795Y (rs1063857) were found to be in strong linkage disequilibrium with each other (r2=1.0), each showing 8 per cent heterozygosity, which also showed moderate degree of linkage disequilibrium with c.2282-42C>A (rs216293) (r2=0.656) (http://www.broadinstitute.org/mpg/snap/ldsearchpw.php). Other polymorphisms which showed significant degree of linkage disequilibrium were c.7288-21C>T(rs3819539) and c.7288-19 C>T(rs3819540)(r2= 0.867). p.N318K has been reported both as mutation and polymorphism; however, the present analysis showed high heterozygosity (11.4%). The novel variant c.8439+207 was checked for its effect on the splicing mechanism by using ALAMUT software21 and was found to be benign.
Due to the complexity of the gene and heterogeneity of mutations, RFLP analysis using intragenic polymorphisms has been an important diagnostic tool to track the affected allele in the family members of severe VWD patients. Rapid automated methods to simultaneously analyze multiple markers by multiplex fluorescent PCR followed by capillary electrophoresis have also been reported to make the diagnosis more cost-effective and less time consuming22. Our study shows the utility of additional polymorphic markers with high heterozygosity frequencies for genetic diagnosis in type 3 VWD.
Acknowledgment
The project was supported by a financial grant from the Council of Scientific and Industrial Research (CSIR), New Delhi.
Conflicts of Interest: None.
References
- The bleeding score predicts clinical outcomes and replacement therapy in adults with von Willebrand disease. Blood. 2014;123:4037-44.
- [Google Scholar]
- The molecular basis of von Willebrand disease: the under investigated, the unexpected and the overlooked. Haematologica. 2011;96:798-800.
- [Google Scholar]
- von Willebrand factor variant database (VWFdb) Available from: www.vwf.group.shef.ac.uk
- [Google Scholar]
- High-throughput molecular diagnosis of von Willebrand disease by next generation sequencing methods. Haematologica. 2012;97:1003-7.
- [Google Scholar]
- Mutation distribution in the von Willebrand factor gene related to the different von Willebrand disease (VWD) types in a cohort of VWD patients. Thromb Haemost. 2012;108:662-71.
- [Google Scholar]
- The mutation spectrum associated with type 3 von Willebrand disease in a cohort of patients from the north west of England. Haemophilia. 2009;15:1048-57.
- [Google Scholar]
- Von Willebrand factor 1 and factor 2 alleles (intron 40) are suitable markers for carrier detection in von Willebrand disease families in the Indian population. Acta Haematol. 2006;115:64-7.
- [Google Scholar]
- A polymorphism of the human von Willebrand factor (vWF) gene with BamHI. Nucleic Acids Res. 1986;14:4697.
- [Google Scholar]
- A second XbaI polymorphic site within the human von Willebrand factor (vWF) gene. Nucleic Acids Res. 1987;15:9099.
- [Google Scholar]
- Two Taq I RFLPs in the human von Willebrand factor gene. Nucleic Acids Res. 1987;15:1347.
- [Google Scholar]
- von Willebrand disease investigated by two novel RFLPs. Br J Haematol. 1988;68:243-8.
- [Google Scholar]
- Sac I RFLP in the human von Willebrand factor gene. Nucleic Acids Res. 1987;15:6766.
- [Google Scholar]
- RsaI RFLP in the human von Willebrand factor gene. Nucleic Acids Res. 1987;15:5909.
- [Google Scholar]
- A Taq I polymorphism in the 5’ region of the von Willebrand factor (vWF) gene. Nucleic Acids Res. 1988;16:2742.
- [Google Scholar]
- An EcoRI polymorphism of the human von Willebrand factor cDNA (vWF) Nucleic Acids Res. 1989;17:6435.
- [Google Scholar]
- Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103-14.
- [Google Scholar]
- Ensembl genome browser 83. Available from: http://genome.ucsc.edu; bioinfo.ut.ee/primer3-0.4.0 www.ensembl.org
- [Google Scholar]
- Molecular characterization of a multiethnic group of 21 patients with type 3 von Willebrand disease. Thromb Haemost. 2000;84:536-40.
- [Google Scholar]
- Available from: http://www.vwf.group.shef.ac.uk/genomicvwf.html
- Available from: www.interactive-biosoftware.com/alamutbatch/features
- Von Willebrand gene tracking by single-tube automated fluorescent analysis of four short tandem repeat polymorphisms. Thromb Haemost. 2005;93:976-81.
- [Google Scholar]




