
Translate this page into:
A simplified multiplex PCR-based typing method for common Salmonella enterica serovars supported by online server-based detection system
Reprint requests: Dr. Probodh Borah, Department of Microbiology, College of Veterinary Science, Assam Agricultural University, Khanapara, Guwahati 781 022, Assam, India e-mail: borahp@vetbifguwahati.ernet.in
-
Received: ,
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
A rapid and simple alternative method is needed to replace the laborious, time-consuming Salmonella serotyping. The objective of the present study was to improve and simplify a previously reported multiplex polymerase chain reaction (PCR)-based method and to create an online server to enable rapid determination of serovars.
Methods:
A method of multiplex PCR-based genome typing (MPGT) was standardized using 59 Salmonella isolates of 31 serovars. Several previously reported primers were modified to obtain a more accurate performance. The screen was separated into four different multiplex reactions distinguishable on standard electrophoresis. A blind study was subsequently performed with 81 isolates of 10 serovars most prevalent in India. Whole genome information from 440 Salmonella isolates was used to confirm the usefulness of this method and concurrence of in silico predictions and PCR results were investigated. A public server (http://www.mpgt-salmonella.res.in) was established for data storage and determination of closest previously observed Salmonella isolates based on obtained MPGT patterns.
Results:
The 16 target genes amplified showed variability in their presence in strains from different serotypes. Hence, identical amplification patterns suggested genetic relatedness of strains and usually identical serological behaviour. The observed absence/presence patterns of genes were converted to an MPGT code. Altogether, 83 different codes were predicted in silico based on the whole genome information of 440 strains. Results confirmed that major serovars usually displayed unique MPGT codes.
Interpretation & conclusions:
The multiplex PCR assay resulted in specific binary codes for isolates from each of the 31 Salmonella serovars tested. The online server allowed the user to compare obtained PCR results with stored previous patterns. Simplicity, speed and cost-effectiveness make this tool useful for quick outbreak management.
Keywords
Molecular typing
multiplex polymerase chain reaction
public database
Salmonella
serovar

Salmonellae are one of the leading causes of community-acquired food-borne bacterial gastroenteritis worldwide. Salmonellae are divided into two species, Salmonella bongori and Salmonella enterica. The species S. enterica is further divided into six subspecies, namely, enterica (I), salamae (II), arizonae (IIIa), diarizonae (IIIb), houtenae (IV) and indica (VI)1. Various strains of S. enterica subsp. enterica are parasitic in humans and warm-blooded animals and are known to be associated with clinical infections2.
Of the known >2500 serovars of Salmonella, about 1500 belong to S. enterica subsp. enterica3. Serotyping of Salmonella strains is conventionally carried out by identification of surface antigens [lipopolysaccharides (LPS), O-antigens] and flagellar antigens (proteins, H-antigens). Although serotyping using the Kauffman-White scheme remains the standard for serovar determination because of its long-standing and widespread use, it has certain deficiencies. Besides being labour-intensive and expensive, serotyping is also time consuming, often requiring three or more days for a highly trained laboratory technician to produce a result4.
Sometimes, atypical expression of surface O or H antigens of an isolate may lead to incomplete or incorrect identification of the serovar. Genomic studies of common clinical serotypes have also revealed a high level of genomic variation amongst isolates of some serovars56. It has been suggested that on the basis of genetic relatedness amongst isolates within the same serovar, a ‘genovar’ (more precisely, ‘genomovar’) classification may be adopted78.
With the increased availability of genomic sequence information, attention has been focused on exploiting this information for finding a molecular strategy of identification of antigenic diversity amongst Salmonella strains, either as a complementary technique or as replacement for conventional serotyping. Approaches include polymerase chain reaction (PCR)-based techniques to determine different O and H antigens910, ribotyping11, pulsed-field gel electrophoresis (PFGE)12, multiplex PCR1113, IS200 analysis14, random amplification of DNA polymorphisms15, multilocus sequence typing1617, matrix-assisted laser desorption ionization-time of flight (MALDI-TOF)-mass spectrometry (MS)18 and DNA microarray analysis19. These typing methods have been reviewed by Al-Mogbel et al20.
Based on the genetic differences amongst the serotypes, Leader et al4 reported the development of a rapid, high-throughput multiplex PCR-based method that was able to discriminate the majority of common serotypes reported in the United States. The use of this typing method, especially in conjunction with serogrouping or PFGE, allowed for serovar determination of Salmonella isolates at a level comparable to that of conventional serotyping, with considerable time and cost savings.
However, as the method involves fluorescent labelling of the products and subsequent detection by capillary electrophoresis, it may not be suitable for use in routine laboratories in developing countries. The present study was undertaken with a view to increase the specificity and sensitivity of this method by modifying some primers and to simplify the protocol by obviating fluorescent labelling and replacing the capillary detection system with conventional gel electrophoresis. The study was further extended by developing a freely accessible online server for storage and expansion of obtained multiplex PCR patterns to facilitate rapid determination of serovars by correlating the patterns with previously deposited patterns.
Material & Methods
A total of 45 Salmonella isolates belonging to 22 different serovars (at least two isolates per serovar) were obtained from the strain collection at University of California (UC), Irvine. Additional 14 S. enterica subsp. enterica isolates of serovars Typhimurium (2), Typhi, Paratyphi A, Virchow, Newport, Schwarzengrund, Enteritidis, Worthington, Gallinarum, Infantis, Idikan, Vridi and Paratyphi B were obtained from the Microbial Type Culture Centre, Chandigarh, and the National Institute of Cholera and Enteric Diseases, Kolkata, India. These strains were used in the code determination phase of the project. For code verification experiments, 81 additional strains (22 Enteritidis, 16 Typhi, 15 Typhimurium, 10 Newport, 8 Weltevreden, 4 Dublin, 2 Litchfield, 2 Gallinarum, 1 Paratyphi B and 1 Virchow) isolated in India from May 2011 to December 2014 from human (28), poultry (23), cattle (18), wild birds (7), pig (2), tiger (1), snake (1) and mouse (1) were tested. The part of the work involving strains (45) from UC, Irvine, including the in silico analysis of genomic sequences (440) was done in the department of Microbiology and Molecular Genetics, University of California, Irvine, USA, between June and November 2010. The part involving Indian isolates (95) including creation and hosting of database was done in College of Veterinary Science, Assam Agricultural University, Guwahati, India, during 2011-2014.
For preliminary standardization of the method, a DNA template from the sequenced strains of S. enterica serovar Typhimurium LT2 (NCBI Bioproject #PRJNA57799), S. enterica serovar Typhi CT18 (PRJNA57793) and S. enterica serovar Enteritidis P125109 (PRJNA31109) were prepared using the Sigma GenElute Bacterial DNA kit (Sigma-Aldrich Corp., St. Louis, MO, USA). These isolates were also tested through colony PCR21, where a colony suspension was boiled in a total volume of 75 μl of ×1 TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) for 20 min at 100°C and then used directly in the multiplex reaction. DNA of the other strains investigated was prepared using the Sigma GenElute Bacterial DNA kit.
Multiplex PCR: Four multiplex-specific 10x primer master mixes were prepared to contain 4 μM of each primer. All PCRs were carried out in a final volume of 25 μl containing 0.5 μl of Taq DNA polymerase (Qiagen Inc., USA), 2.5 μl of the respective 10x primer master mix and 1 μl of template DNA (either genomic DNA or simple boiled culture preparations in TE, pH 8). Thermocycling parameters were 94°C for 3 min; 30 cycles of 94°C for 15 sec, 50°C for 30 sec and 72°C for 25 sec; and 72°C for 5 min. Control reaction mixtures containing no template or genomic DNA from Salmonella serovar Typhimurium LT2, serovar Typhi CT18 or serovar Enteritidis PT4 were included in each sample run.
Gel electrophoresis and code determination: The PCR products were separated by gel electrophoresis on a 2 per cent (w/v) agarose gel. The resulting product was manually scored, based on the presence of a PCR product that corresponded to the predicted amplicon size. Control reactions with DNA from Salmonella serovars Typhimurium, Typhi or Enteritidis were employed to verify expected amplicon sizes. A multiplex PCR genome typing (MPGT) code was determined for each tested isolate as follows: all amplicons included in each of the multiplexes (M-1 through M-4) were arranged in descending order of their corresponding product size. The internal control, Salmonella-specific region STM1608, resulted in a visible 63bp amplicon in every Salmonella isolate and was included in every multiplex. This control reaction was not integrated into the code. For the other amplicons, a successful PCR product was then indicated by 1, a failure to amplify by 0. The resulting code consisted of four blocks of binary numbers, separated by hyphens, corresponding to the four multiplexes used in this identification scheme (M1-M2-M3-M4). Each digit corresponded to a gene in these multiplexes.
Optimization of primer sequences and concentrations: For modification of the PCR regimen employed by Leader et al4, all original 16 target genes were retained, but the fluorescent-labelled universal probe sequence tag was eliminated from each primer. The sequences of 18 of the 32 original primers employed in that study4 were preserved. Performance of the remaining 14 primers (STM1608R, STM0839F, STM0839R, STM1350R, STM4538F, STM0716F, STM0716R, STM3845F, STM3845R, STM4525F, STM4525R, STM3518F, STY0311F and STY0311R) was found to be suboptimal in the present study. These primers were modified, either through shifting of the target sequence by two or three nucleotides or through an entirely new design using Primer-BLAST against the previously reported target gene sequences. The Table lists all primers used in the modified protocol.
Based on their expected product sizes, the primer pairs were divided into four separate multiplex sets (M-1, M-2, M-3 and M-4, Table), allowing easy separation of the amplified products during standard gel electrophoresis. Primers directed towards a universal Salmonella- specific region (STM1608) were incorporated in each of the multiplex groups as internal DNA amplification control.
The observed absence/presence patterns of the multiplexed PCR reactions were converted to an MPGT code. The identical amplification patterns suggested relatedness of strains and usually an identical serological behaviour. Subsequently, a more in-depth blind study was performed for the most important and/or common isolates prevalent today (code verification). For this, 40 isolates belonging to five different serovars (Typhimurium, Typhi, Enteritidis, Gallinarum and Dublin) were tested.
In silico determination of multiplex PCR-based genome typing (MPGT) codes for Salmonella genomes: A total of 440 Salmonella genome sequences available as NCBI BioProjects (https://www.ncbi.nlm.nih.gov/bioproject) were interrogated for determination of their theoretical MPGT codes. These sequences included at least 85 different serovars and isolates from all six S. enterica subspecies. BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was employed to investigate primer hit locations in these genomes, using relaxed similarity parameters (minimum bit score=20 and minimum word size=8) and absence or presence of the desired PCR product was inferred.
Server-based multiplex PCR-based genome typing (MPGT) pattern comparisons: An online database and server supported by a database (http://www.mpgt-salmonella.res.in) were developed for quick and simple comparison of the multiplex PCR pattern obtained from an unknown test strain to previously observed patterns, with the inference of close relationships. The implementation of the database was carried out on the WAMP (Windows, Apache, MySQL, PHP) platform. The user connects to the MySQL-MPGT code database through a web browser.
The database consisted of reference data pertaining to 31 clinically important Salmonella serovars, their MPGT codes and the corresponding antigenic properties. In addition, the database also included predicted MPGT codes of at least 85 Salmonella serovars based on in silico analysis of 440 Salmonella genome sequences available as NCBI BioProjects. After entry of vital strain metadata such as location, host and date of its isolation and associated clinical symptoms, the user simply indicates the status of the targeted amplicons by clicking on the corresponding text boxes against the specific products (Fig. 1). All submissions made by a registered user are stored in the server for future references.

- Screenshot of the data entry window in the multiplex PCR-based genome typing code server.
If the user's submitted results do not match any existing MPGT codes in the database, the five closest matches (serovars and their MPGT codes) will be displayed, based on a scoring matrix developed for this purpose. Briefly, the MPGT codes of user inputs and those in the database are split from strings into characters and fetched into arrays. After that, the elements of the users’ arrays are matched 1:1 (proceeding from index zero to the last element of the array) with MPGT codes of each of the serovars fetched earlier using arrays. If the values match, the score is set to 1, if not, it is 0. The sum of the scores is calculated. Finally, the percentage of total score with respect to the total length of the MPGT codes is calculated. If the score matches 100 per cent, then the server displays the matched serovar along with five nearest matching serovars; otherwise, it shows the nearest five serovars with their matching percentages. The users are able to view not only the serotype of the closest match but also the associated metadata.
Results
The protocol standardized for the multiplex PCR reactions using one standard strain each of S. Typhimurium (LT2), S. Typhi (CT18) and S. Enteritidis (PT4) yielded distinct band patterns of the target genes with expected product sizes on agarose gel electrophoresis (Fig. 2).
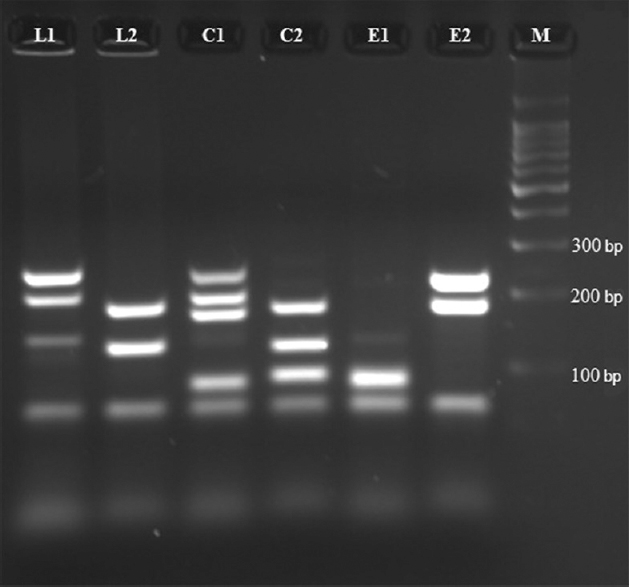
- Amplicons of different sizes obtained by multiplex polymerase chain reaction. L1 & L2, Multiplex (M) 1 and M2 for S. Typhimurium (LT2 strain); C1 & C2, M1 and M2 for S. Typhi (CT18 strain); E1 & E2, M1 and M2 for S. Enteritidis (PT4 strain). M: 100 bp DNA ladder.
Overall, 83 different MPGT codes were predicted for the 440 strains. Results confirmed that the major serovars usually resulted in separate and unique MPGT codes, exemplified by the Typhimurium-specific code (11010-0110-111-101), found in 22 Typhimurium isolates, including the monophasic 4,5,12,i,- variant, the Paratyphi A/Sendai code (00101-0110-100-010), found in four isolates and a distinct code for Enteritidis isolates (00001-1100-011-101), detected in 84 isolates. For Typhi, 14 of the 15 isolates resulted in the same distinct MPGT code (01101-0111-000-010), with only Typhi Ty2 displaying a single-digit predicted difference (01001-0111-000-010). Occasionally, closely related strains of different serovars resulted in identical predicted MPGT codes (for example, strains of the serovars Gallinarum and Dublin, code 00001-0100-011-101, Kentucky and Hadar, code 01000-0100-111-001), and some serovars displayed a variety of codes (such as Montevideo, where 41 of the 53 isolates displayed a prominent code 00110-0100-010-001, but the remaining displayed four variations of MPGT codes). Those serovars may be of polyphyletic origin as previously observed22. However, limited utility of the multiplex PCR approach was observed for the distinction of isolates from subspecies other than subspecies I, as many of these failed to result in a reasonable BLAST hit for most of the primers. Most of these interrogated genome sequences were only high-quality drafts consisting of multiple contigs, so the in silico MPGT code determination for these genomes was preliminary.
PCR-derived MPGT codes for common clinical human and animal Salmonella serovars: Initially, 63 isolates were used to determine wet (as opposed to in silico) MPGT codes for 31 Salmonella serovars including serovars Typhimurium, Typhi and Enteritidis. For 16 of the 31 serovars tested, a unique MPGT code was produced. Isolates of Salmonella serovars Dublin, Gallinarum var. Pullorum, Heidelberg, Kentucky, Paratyphi A, Paratyphi C, Typhi and Weltevreden each produced two MPGT codes. As a notable difference to the in silico analysis, Typhimurium isolates also produced two codes, differing by the status of STM3518. Duplicate codes were also obtained for Salmonella bongori and S. enterica subsp. II, IIIa, IIIb, IV and VI. All serovars screened in the panel resulted in distinct MPGT code identifiers.
In the blind study, 77 (95.06%) of the 81 tested strains resulted in the expected MPGT code identifiers, suggesting a relatively high reproducibility and specificity of the multiplex PCR approach. For Salmonella Typhimurium, 13 of 15 isolates resulted in an MPGT code that corresponded to the code produced by Typhimurium isolates during code determination experiments. Amongst these 13 isolates, seven showed the specific in silico-predicted MPGT code (11010-0110-111-101) and the remaining six displayed the alternative code (11010-0110-111-001), which was deficient of the gene STM3518. However, two of the 15 tested strains that had been stocked as Typhimurium resulted in an MPGT code (00001-1100-011-101) produced by Enteritidis strains during code determination (a code that differs from expected Typhimurium codes by at least 8 out of 16 sites), indicating a high likelihood of mistyping or misrecording of these strains. Twenty one of 22 tested isolates of Salmonella Enteritidis resulted in MPGT codes indicative of serovar Enteritidis (00001-1100-011-101). The remaining isolate tested displayed a MPGT code indicative of serovar Gallinarum in our code determination tests (00000-0110-101-001), suggesting a typing or recording error. All 16 isolates of the human host-specific serovar S. Typhi were correctly identified by the multiplex PCR method. Nine of these showed the typical code (11101-0111-000-010), whereas seven displayed a code devoid of gene STM4525 (01101-0111-000-010), the prevalent Typhi code identified in the in silico analysis. All eight isolates of S. Weltevreden were correctly identified by the method, of which four each showed the alternative codes (00110-0110-110-001 and 10110-0111-110-001). Three of the four tested isolates of Salmonella Dublin resulted in the expected MPGT code (00001-0100-011-101), and the remaining one resulted in an MPGT code identical to a code previously obtained from a Typhimurium isolate (11010-0110-111-101, a difference in 6 sites), underlining utility of the MPGT screen to identify strains that had been misrecorded. Both tested isolates of Salmonella Gallinarum resulted in their expected MPGT code (00000-0110-101-001). Both isolates of S. Litchfield (11001-0110-111-001) and the single isolate each of S. Paratyphi B (11000-0110-110-001) and S. Virchow (01000-0110-101-001) tested in blind study showed codes typical of the serovars as determined earlier by code determination with standard strains.
Comparison of PCR results with in silico predictions: For 13 of the 31 tested serotypes investigated in the code determination experiments, obtained patterns concurred entirely with codes predicted by the in silico analysis for isolates of the same serotype. This lower than expected concordance prompted us to compare in silico and laboratory patterns obtained for the 28 strains where both analyses had been performed. Overall, in silico analysis correctly predicted the status of 381/420 amplification products (90.7%). All but four of the discrepancies were cases where the in silico analysis failed to predict an obtained PCR product, suggesting either PCR product generation based on weak sequence similarities that cannot be picked up by a simple BLAST search or, alternatively, unexpected product generation due to primer crosstalk.
Discussion
Although conventional serotyping offers a reliable method for differentiating Salmonella strains, it is a time-consuming process. Among other methods, PFGE is highly discriminative for Salmonella, but it is expensive and time consuming. Further, its standardization, analysis and comparison of restriction profiles require effort20. Ribotyping is a labour-intensive and time-consuming technique, analysis may take in some cases 4-5 days to complete23. IS200 profiling has been found to have low discriminatory power24. The PCR-based methods such as RAPD lack the reproducibility between laboratories25. Similarly, the results of ERIC-PCR are usually less discriminatory26. MLST does not provide the fine resolution needed for outbreak analysis and short-term epidemiology20. Although Matrix-assisted laser desorption/ionization Time-of-flight Mass Spectrometry (MALDI-TOF-MS) can be a rapid method for typing, laborious control of the sample preparation and optimization of testing parameters are crucial for strain typing with this method, which is not practical in clinical laboratories27.
The combination of the four multiplex PCRs reported here resulted in specific binary codes for isolates from every one of 31 serovars tested in code determination experiments including isolates from the most common and/or most severe serovars (Typhimurium, Enteritidis, Typhi and Paratyphi A). For 16 of the 31 serovars tested, a single MPGT code was produced for each serovar.
The code verification experiments correctly identified the closest relative within the expected serovar in >95 per cent of the examined Salmonella strains. This imperfect concordance level may, in part, stem from possible cross-reaction of the primers in the multiplex combinations with each other or certain target DNA sequences. This was supported by the fact that the in silico analysis correlated not entirely with the obtained laboratory PCR results and failed to predict a few observed amplified products. However, some of the inconsistencies may also be caused by incorrect conventional serotyping based on human error or the quality of the antiserum lots28 and by incorrect recording of stocked isolates. Finally, it cannot be assumed that strains will result in patterns that have been observed for other isolates of the same serovar. It is known that serotype is not always a measure of genetic relatedness29, and therefore, a DNA-based method like this multiplex PCR approach may initially miscategorize observed amplification patterns. However, as the server is constantly enriched by more observations, the last type of discrepancy will become less relevant.
For most of the strains tested in the present study, the gene profiles corresponded to those previously reported4 for all 16 loci. However, some discrepancies were observed. Unlike the results reported by Leader et al4, gene STM4525 was detected in our multiplex PCR test for two isolates of Typhi and all three Paratyphi A isolates tested. Our primers for this target gene differed from those originally reported in that study4. The in silico analysis suggested this gene to be absent from Typhi and Paratyphi A isolates, and amplification of this product by PCR may, therefore, constitute a template-specific artefact. Similarly, STY2349 was inexplicably but repeatedly amplified from single isolates of Choleraesuis, Kentucky and Virchow, and STM0839 was detected in one Heidelberg strain.
The major advantages of using the present multiplex PCR to determine Salmonella relatedness were its speed, simplicity and relative accuracy. An isolate could be typed within 1.5 to two hours with a reasonable level of accuracy, a clear advantage over the conventional serotyping method. Although conventional serotyping is considered the gold standard for Salmonella identification, it occasionally demonstrates inconsistent results30.
An additional benefit of the multiplex PCR method is the possibility to determine a molecular profile for those strains that cannot be serotyped (i.e., those that do not express antigen or have an LPS defect rendering them untypable). This occurs for 5-8 per cent of all strains routinely tested in laboratories13. Compared to other methods such as PFGE, ribotyping and MALDI-TOF-MS, the present method is easier to perform, less expensive and less time consuming. However, this method needs further validation against type cultures of other common clinical serovars and a large number of field isolates. Users can submit an obtained novel code to the MPGT database either as a new serovar (known or unknown) or as an alternative code for an existing serovar. Through user input, the database is expected to expand over time with continuous incorporation of data pertaining to more and more strains and serovars.
In conclusion, the present multiplex-PCR-based method supported by the freely-accessible online server provided a simple, rapid and reasonably accurate alternative to conventional serotyping for determination of serovars in clinical isolates of Salmonella. As more and more MPGT codes get incorporated into the database, accuracy of its closest relative predictions will improve, and its utility vastly expanded. The simplicity of the method allowed this crude molecular typing of common clinical Salmonella isolates to be performed in any laboratory without much sophistication. This method could be useful for quick outbreak management.
Acknowledgment
The authors acknowledge the Department of Biotechnology, Govt. of India, for providing necessary financial support in the form of DBT Overseas Associateship provided to the first author and subsequent funding in the form of DBT-NER Twinning Project (BT/375/NE/TBP/2012) to carry out the study.
Conflicts of Interest: None.
References
- Evolutionary genomics of Salmonella enterica subspecies. MBio. 2013;4 (pii) : E00579-12
- [Google Scholar]
- Supplement 1997 (no.41) to the Kauffmann-White scheme. Res Microbiol. 1998;149:601-4.
- [Google Scholar]
- Antigenic formulae of the Salmonella serovars. In: WHO Collaborating Centre for Reference and Research on Salmonella (9th ed). Paris, France: Institut Pasteur; 2007. p. :1-166.
- [Google Scholar]
- High-throughput molecular determination of Salmonella enterica serovars by use of multiplex PCR and capillary electrophoresis analysis. J Clin Microbiol. 2009;47:1290-9.
- [Google Scholar]
- Comparative genomics of closely related salmonellae. Trends Microbiol. 2002;10:94-9.
- [Google Scholar]
- Comparison of Salmonella enterica serovar typhimurium LT2 and non-LT2 salmonella genomic sequences, and genotyping of salmonellae by using PCR. Appl Environ Microbiol. 2006;72:6142-51.
- [Google Scholar]
- Differences in gene content between Salmonella enterica serovar Enteritidis isolates and comparison to closely related serovars Gallinarum and Dublin. J Bacteriol. 2005;187:6545-55.
- [Google Scholar]
- Salmonella serovar identification using PCR-based detection of gene presence and absence. J Clin Microbiol. 2008;46:2581-9.
- [Google Scholar]
- Multiplex PCR-based detection and identification of the most common Salmonella second-phase flagellar antigens. Res Microbiol. 2002;153:107-13.
- [Google Scholar]
- Multiplex PCR for distinguishing the most common phase-1 flagellar antigens of Salmonella spp. J Clin Microbiol. 2004;42:2581-6.
- [Google Scholar]
- Use of ribotyping for characterization of Salmonella serotypes. J Clin Microbiol. 1993;31:233-7.
- [Google Scholar]
- Molecular Typing of Salmonella Strains Isolated from Food, Feed and Animals: State of Play and Standard Operating Procedures for Pulsed Field Gel Electrophoresis (PFGE) and Multiple-Locus Variable Number Tandem Repeat Analysis (MLVA) Typing, Profiles Interpretation and Curation. 2014. EFSA Supporting Publication. EN703:1-74. Available from: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2014.EN-703/epdf
- [Google Scholar]
- Development of multiplex PCR for rapid identification of four Salmonella serovars most commonly isolated in Japan. Southeast Asian J Trop Med Public Health. 2014;45:654-61.
- [Google Scholar]
- Application of ribotyping and IS200 fingerprinting to distinguish the five Salmonella serotype O6,7:c:1,5 groups: Choleraesuis sensu stricto, Choleraesuis var. Kunzendorf, Choleraesuis var. Decatur, Paratyphi C, and Typhisuis. Epidemiol Infect. 1999;123:37-46.
- [Google Scholar]
- Population structure of Salmonella investigated by amplified fragment length polymorphism. J Appl Microbiol. 2004;97:566-73.
- [Google Scholar]
- Multilocus sequence typing for characterization of clinical and environmental salmonella strains. J Clin Microbiol. 2002;40:1626-35.
- [Google Scholar]
- Multilocus sequence typing as a replacement for serotyping in Salmonella enterica. PLoS Pathog. 2012;8:e1002776.
- [Google Scholar]
- Rapid classification and identification of salmonellae at the species and subspecies levels by whole-cell matrix-assisted laser desorption ionization-time of flight mass spectrometry. Appl Environ Microbiol. 2008;74:7767-78.
- [Google Scholar]
- Microarray for molecular typing of Salmonella enterica serovars. Mol Cell Probes. 2008;22:238-43.
- [Google Scholar]
- A review on typing of non-typhoidal Salmonella (NTS) J Pure Appl Microbiol. 2015;9:461-8.
- [Google Scholar]
- Reliable means of diagnosis and serovar determination of blood-borne Salmonella strains: Quick PCR amplification of unique genomic loci by novel primer sets. J Clin Microbiol. 2009;47:2435-41.
- [Google Scholar]
- Genetic boundaries to delineate the typhoid agent and other Salmonella serotypes into distinct natural lineages. Genomics. 2013;102:331-7.
- [Google Scholar]
- Comparison of ribotyping and arbitrarily primed PCR for molecular typing of Salmonella enterica and relationships between strains on the basis of these molecular markers. J Appl Microbiol. 1998;85:933-40.
- [Google Scholar]
- Molecular typing of Salmonella typhi strains from Dhaka (Bangladesh) and development of DNA probes identifying plasmid-encoded multidrug-resistant isolates. J Clin Microbiol. 1996;34:1373-9.
- [Google Scholar]
- Factors affecting reproducibility of random amplified polymorphic DNA fingerprinting. Res Microbiol. 1993;144:373-9.
- [Google Scholar]
- Molecular typing methodologies for microbial source tracking and epidemiological investigations of Gram-negative bacterial foodborne pathogens. Infect Genet Evol. 2009;9:430-40.
- [Google Scholar]
- Rapid screening of epidemiologically important Salmonella enterica subsp. enterica serovars by whole-cell matrix-assisted laser desorption ionization-time of flight mass spectrometry. Appl Environ Microbiol. 2011;77:4136-46.
- [Google Scholar]
- Development and evaluation of a multiplex polymerase chain reaction assay to identify Salmonella serogroups and serotypes. Diagn Microbiol Infect Dis. 2009;65:327-30.
- [Google Scholar]
- Genomic comparison of Salmonella enterica serovars and Salmonella bongori by use of an S. enterica serovar typhimurium DNA microarray. J Bacteriol. 2003;185:553-63.
- [Google Scholar]
- Association of Public Health Laboratories. Salmonella serotyping in US public health laboratories. Silver Spring, MD: Association of Public Health Laboratories; 2014. p. :4.
- [Google Scholar]




