
Translate this page into:
A novel ready-to-use dry-reagent polymerase chain reaction for detection of Escherichia coli & Shigella species
For correspondence: Dr Shama Bhat, Bhat Biotech India Pvt. Ltd., 11-A, 4th Cross, Veerasandra Industrial Area, Electronic City Phase II, Bengaluru 560 100, Karnataka, India e-mail: bhatbiotech@gmail.com
-
Received: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
Polymerase chain reaction (PCR) has wide acceptance for rapid identification of pathogens and also for diagnosis of infectious conditions. However, because of economic and expertise constraints, a majority of small or peripheral laboratories do not use PCR. The objective of the present study was to develop a dry-reagent PCR assay as an alternative to conventional PCR to assess its applicability in routine laboratory practice using malB gene for identification of Escherichia coli as a model.
Methods:
A total of 184 isolates were selected for the study comprising clinical isolates of E. coli and non-E. coli including Shigella sp. and a few other control strains. The DNA was isolated from all the isolates. The isolated DNA as well as the overnight grown bacterial cultures were subjected to both conventional wet PCR and dry-reagent PCR.
Results:
The genomic DNA isolated from E. coli showed amplification of malB gene in both conventional wet and dry-reagent PCR and the band was observed at 491 bp. In dry-reagent PCR, the overnight grown E. coli cells also showed positive result. The non-E. coli strains other than Shigella sp. showed negative in both conventional wet and dry-reagent PCR. Shigella sp. showed positive in both conventional wet and dry-reagent PCR.
Interpretation & conclusions:
Considering the elimination of genomic DNA isolation step, and similar results with the conventional wet PCR, dry-reagent PCR may be a good alternative for the conventional wet PCR.
Keywords
ABC transporter
dry-reagent PCR
Escherichia coli
malB gene
maltose/maltodextrin transporter ATP-binding gene
pathogens
Shigella sp.

Escherichia coli a commensal Gram-negative bacillus, has various pathotypes causing gastrointestinal and most other pyogenic infections such as wound infections, septicaemia and meningitis. E. coli is the most common cause of urinary tract infections and now is placed in the list of important hospital pathogens1. It is routinely identified by standard biochemical and physiological tests. PCR assays though are available for molecular identification of E. coli2345, these are not commonly used by the small laboratories because of high cost of testing and lack of expertise. Therefore, an effort was made to develop a dry-reagent mix for PCR targeting malB gene commonly used to identify E. coli45. The use of dry-reagent mix with an enzyme resistant to PCR inhibitors has helped us to eliminate the step of DNA extraction from bacterial isolates. This feature enabled the use of this assay directly on bacterial suspensions as well as on isolated DNA for the identification of E. coli. The specificity of this test was assessed using a number of bacterial isolates other than E. coli (non-E. coli). Here, we report the development of optimized ‘ready-to-use’ dry-reagent mix dispensed in individual PCR tubes.
Material & Methods
The study was conducted at Bhat Biotech India Pvt. Ltd., Bengaluru, and department of Microbiology, SDM College of Medical Sciences and Hospital, Dharwad, India, from June 2015 to June 2017. All clinical isolates were identified to species level by commonly used conventional methods6. A total of 184 isolates were used in this study of which 104 were phenotypically confirmed Escherichia coli, 73 phenotypically confirmed non-E. coli and seven control strains including two E. coli and five non-E. coli (Table).
| Name of the organism | Number of isolates tested |
|---|---|
| Gram-negative | |
| Acinetobacter sp. | 3 |
| A. baumannii | 3 |
| Citrobacter diversus | 1 |
| C. freundii | 3 |
| Enterobacter sp. | 1 |
| E. cloacae | 1 |
| Klebsiella pneumoniae | 4 |
| Proteus mirabilis | 4 |
| P. vulgaris | 3 |
| Pseudomonas sp. | 1 |
| P. aeruginosa | 4 |
| Salmonella sp. | 1 |
| S. Typhi | 7 |
| Shigella boydii | 2 |
| S. dysenteriae | 2 |
| S. flexneri | 5 |
| S. sonnei | 4 |
| Vibrio cholerae | 2 |
| Staphylococcus aureus | 3 |
| Gram-negative | |
| Coagulase-negative Staphylococci | 4 |
| Methicillin-resistant Staphylococci | 4 |
| Streptococcus sp. | 3 |
| Enterococcus sp. | 4 |
| E. faecalis | 4 |
| Gram-negative | |
| ATCC Escherichia coli 25923 | 1 |
| E. coli DH5α | 1 |
| K. pneumoniae 600703 | 1 |
| ATCC P. aeruginosa 27853 | 1 |
| Gram-positive | |
| ATCC Enterococcus faecalis 29212 | 1 |
| ATCC S. aureus 25922 | 1 |
| Acid fast | |
| Mycobacterium tuberculosis H37Rv | 1 |
Bacterial genomic DNA isolation: A well-isolated single colony of the isolate from brain-heart infusion agar plates was inoculated into 1 ml of sterile Luria-Bertani (LB) broth (HiMedia, Mumbai) and incubated overnight at 37°C. The genomic DNA from all the isolates was extracted using standard protocol7. The eluted DNA was used for both conventional and dry-reagent PCR assays.
Primer selection and PCR conditions: Primers designed for this study were selected from E. coli (accession number LT615379) targeting 491 bp amplicon of malB, sugar ABC transporter and maltose/maltodextrin import ATP-binding protein malK genes. The sequences and location of forward and reverse primers in E. coli chosen were as follows, forward primer 5'-GATGCGTGCACCTGTTTTTA-3’ (4242866 - 4242885 bp) and reverse primer 5'-ACACCACGAATTCACCTTCA-3’ (4243337 - 4243356 bp) (Sigma-Aldrich, USA).
Conventional PCR: The DNA isolated from all 184 bacterial isolates was tested with conventional wet-reagent mix in PCR8. PCR reaction mix was prepared to a final volume of 50 μl containing 32 μl of nuclease-free water, 10 μl of 5X PCR reaction buffer, 2 μl each of 1 μM forward and reverse primer, 1 μl of 10 mM deoxyribonucleotide triphosphates (dNTPs), 1 μl of Taq DNA polymerase (2.5 units/μl) (Fermentas, Vilnius, Lithuania) and 2 μl of isolated DNA as template. PCR reaction mix was short spun and mixed well. The PCR amplification was performed in DNAmp thermocycler (Bhat Biotech India Pvt. Ltd., Bengaluru) and QB-96 (Quanta Biotech, UK) thermocycler. The thermocycling programme was initiated at 97°C for three minutes, followed by 35 cycles comprising denaturation at 94°C for 45 sec, annealing at 57°C for 45 sec, extension at 72°C for 45 sec and final extension at 72°C for 10 min. The PCR products and 1 kb DNA reference ladder (Fermentas, Vilnius, Lithuania) were resolved in 1.5 per cent agarose gel and viewed under ultraviolet (UV) transilluminator (Zenith Engineers, Agra) and photographed.
Dry-reagent mix PCR: The dry-reagent mix PCR was tested with the DNA isolated from all the 184 isolates and also directly with bacterial isolates including test and control. The DNA extracted from the bacterial isolates was used as template, and the test was performed as per the protocol suggested. To the dry PCR tube, 2 μl of template DNA and 48 μl of PCR grade water were added and spun down. The amplification was performed in a QB-96 thermocycler. The PCR conditions were same as that of conventional PCR. The amplified PCR products were analyzed by agarose gel electrophoresis and the DNA was visualized in UV transilluminator and photographed.
All the procedures for dry PCR were the same as above except for the template. Here, 5 μl of bacterial culture in LB broth was added to the dry-reagent PCR tube which served as template followed by 45 μl of PCR grade water and was short spun. The advantage being elimination of DNA extraction step from bacterial culture and the bacterial suspension was directly added to the dry-reagent PCR tube. The designed primers were verified with NCBI BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and the multiple sequence alignment was performed with Multalin version 5.4.19 to confirm the homology between E. coli (accession no. LT615379) and Shigella sp. (accession no. CP014768).
Results
The study group included 177 isolates from clinical samples and seven control strains belonging to different species. Of these isolates, 106 (104 clinical isolates and 2 control strains) were phenotypically confirmed as E. coli and 78 (73 clinical isolates and 5 control strains) were non-E. coli bacterial isolates (Table). The genomic DNA isolated from these 184 isolates was used as template for conventional wet PCR. The conventional PCR was considered as the gold standard for determining sensitivity and specificity of our primer and dry-reagent master mix. All the 104 E. coli isolates and two control strains amplified the target by both conventional wet PCR and dry-reagent PCR (Fig. 1A & B), while 60 non-E. coli clinical isolates and five controls did not show amplification. All the 13 different species of Shigella showed the presence of the target DNA sequence of 491 bp in both conventional wet-reagent and dry PCR (Fig. 2A & B). It was found that malE gene, ABC transporter and maltose/maltodextrin transporter ATP-binding gene of Shigella showed high sequence similarity with malB sequence of E. coli (Fig. 3).
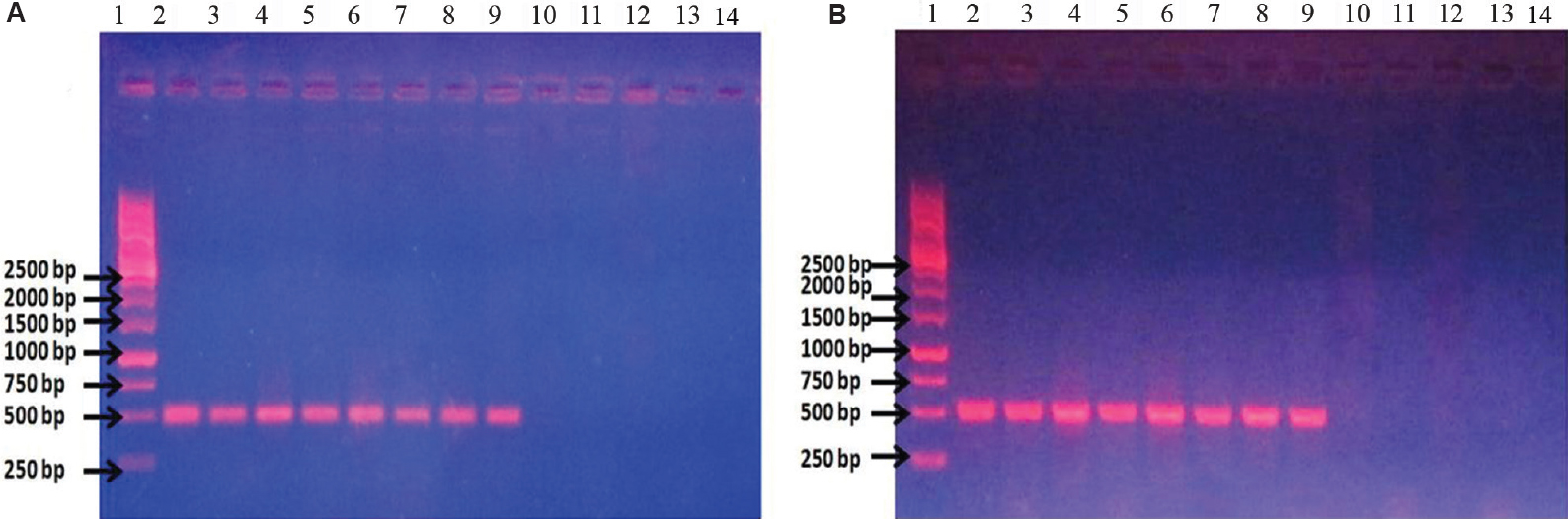
- (A) Conventional wet mix PCR with malB primers. Lane 1: ladder; lanes 2 and 3: Escherichia coli DH5α DNA; lanes 4 and 5: uropathogenic E. coli (UPEC) U110 DNA; lanes 6 and 7: UPEC U111 DNA; lanes 8 and 9: UPEC U310 DNA; lanes 10 and 11: Pseudomonas sp. DNA; lanes 12 and 13: Klebsiella pneumoniae DNA; lane 14: blank. (B) Dry-reagent PCR with malB primers. lane 1: ladder; lane 2: E. coli DH5α DNA; lane 3: E. coli DH5α culture; lane 4: UPEC U110 DNA; lane 5: UPEC U110 culture; lane 6: UPEC U111 DNA; lane 7: UPEC U111 culture; lane 8: UPEC U310 DNA; lane 9: UPEC U310 culture; lane 10: Pseudomonas sp. DNA; lane 11: Pseudomonas sp. culture; lane 12: K. pneumoniae DNA; lane 13: K. pneumoniae culture; lane 14: blank.
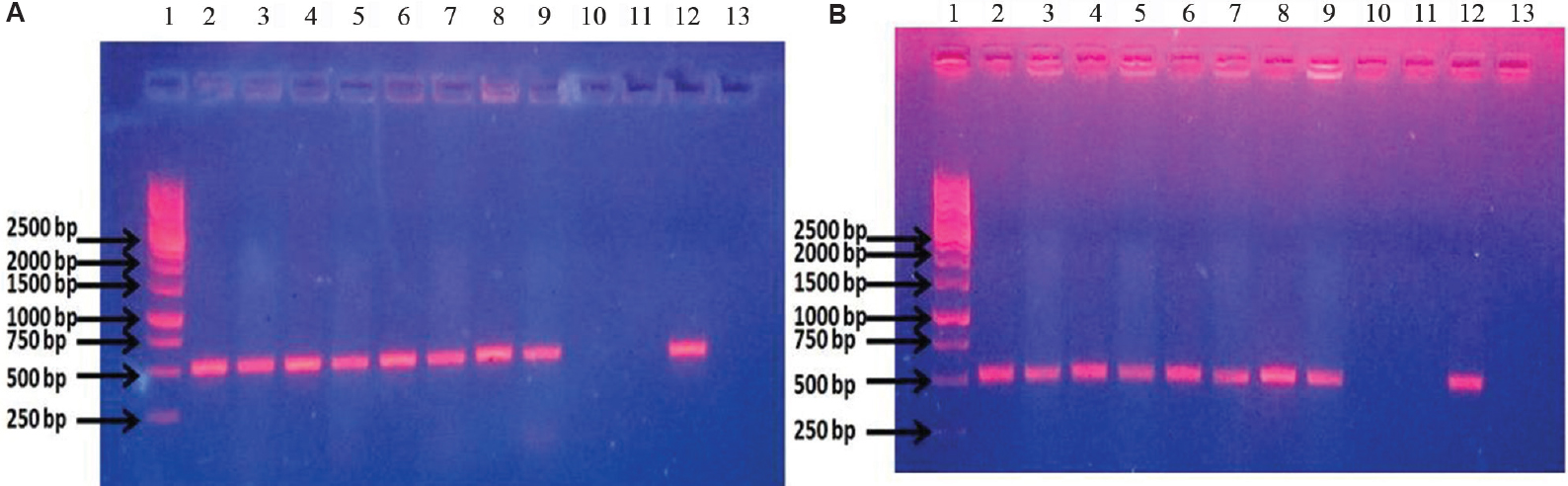
- (A) Conventional wet mix PCR with malB primers. Lane 1: ladder; lanes 2 and 3: Shigella flexneri DNA; lanes 4 and 5: S. sonnei DNA; lanes 6 and 7: clinical isolate of Shigella DNA; lanes 8 and 9: uropathogenic Escherichia coli (UPEC) U110 DNA; lanes 10 and 11: Klebsiella pneumoniae DNA; lane 12: malB clone as template for positive control; lane 13: blank. (B) Dry-reagent PCR with malB primers. Lane 1: ladder; lane 2: S. flexneri DNA; lane 3: S. flexneri culture; lane 4: S. sonnei DNA; lane 5: S. sonnei culture; lane 6: clinical isolate of Shigella DNA; lane 7: clinical isolate of Shigella culture; lane 8: UPEC U110 DNA; lane 9: UPEC U110 culture; lane 10: K. pneumoniae DNA; lane 11: K. pneumoniae culture; lane 12: malB clone as template for positive control; lane 13: blank.
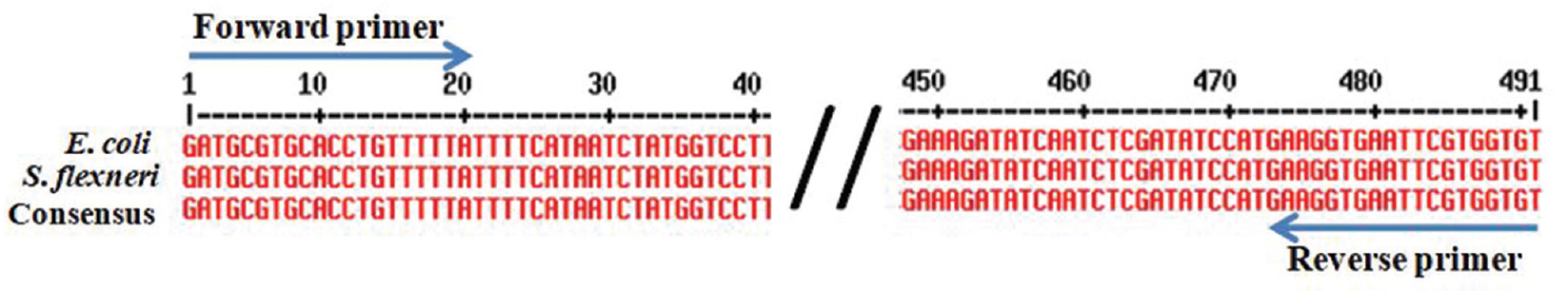
- Multiple sequence alignment of Escherichia coli (accession no. LT615379) and Shigella sp. (accession no. CP014768) and their consensus sequence. Arrow marks indicate the forward and reverse primer.
Discussion
Dry-reagent PCR is not a popular technique in spite of having number of merits. Dry-reagent PCR has been successfully employed for the rapid detection of Mycobacterium species, M. ulcerans, Salmonella Typhi, Vibrio cholerae, Clostridium sp., Staphylococcus sp., Acinetobacter sp. and Yersinia pestis1011121314151617181920. Dry-reagent mix can also be used for quantitative PCR2122, real-time reverse transcription PCR23 and isothermal PCR24. Carbohydrate polymers have been used to stabilize PCR mix which forms glassy matrices and provide room temperature stability25 without compromise or decline in their efficacy even after a year of storage at 20°C26. Several techniques of freeze-drying PCR reagents providing phenomenal stability have been used by different workers101214152021222324.
The dry-reagent PCR assay for malB gene showed 100 per cent agreement with conventional PCR assay. The 13 strains of Shigella were also identified in the PCR. This gene is conserved across diverse lineages of E. coli and is not shared by other Gram-negative bacteria except Shigella sp., based on BLAST analysis. We considered this region to be appropriate because of its conservation, as well as the rarity of Shigella as a cause of extraintestinal infections5. Genome comparisons showed that Shigella shared a common backbone sequence with E. coli27, and the nucleotide homology was nearly 90 per cent28. Molecular evolutionary studies have shown that enteroinvasive E. coli (EIEC) and Shigella can be regarded as a single pathovar of E. coli29.
The lyophilization was not used in our study to convert the reaction mixture in dry format, thus reducing cost of freeze-drying. The significant modification was the elimination of DNA extraction step. DNA extraction is critical for the success of any molecular assay. Poor-quality DNA often gives erroneous results. Most laboratories use either column-based or phenol:chloroform-based DNA extraction. Column-based DNA extraction is commercially available; however, it is expensive. The phenol:chloroform extraction though cheap is time-consuming and hazardous. Dry-reagent PCR master mix was user-friendly by further reducing the stages of storing and thawing of reagents before use and preparation of master/reaction mixture. Therefore, chances of activity loss and contamination of reagents were eliminated.
In conclusion, the dry-reagent PCR developed in this study was speedy, less cumbersome and user-friendly without compromising the sensitivity and specificity. As there is no need to establish a dedicated PCR laboratory, peripheral laboratories may also use this molecular assay.
Financial support & sponsorship: Authors acknowledge the financial assistance provided by the Bhat Biotech India Pvt. Ltd., Bengaluru.
Conflicts of Interest: None.
References
- Hospital-acquired infections due to Gram-negative bacteria. N Engl J Med. 2010;362:1804-13.
- [Google Scholar]
- Improved detection of enterotoxigenic Escherichia coli among patients with travelers’ diarrhea, by use of the polymerase chain reaction technique. J Infect Dis. 1999;180:2053-5.
- [Google Scholar]
- PCR detection of Escherichia coli O157:H7 directly from stools: Evaluation of commercial extraction methods for purifying fecal DNA. J Clin Microbiol. 2000;38:4108-13.
- [Google Scholar]
- Detection of Escherichia coli and identification of enterotoxigenic strains by primer-directed enzymatic amplification of specific DNA sequences. Int J Food Microbiol. 1991;12:339-51.
- [Google Scholar]
- Loop-mediated isothermal amplification assay for rapid detection of common strains of Escherichia coli. J Clin Microbiol. 2008;46:2800-4.
- [Google Scholar]
- Bergey's manual of determinative bacteriology (6th ed). Baltimore: Williams & Wilkins; 1974.
- Molecular cloning. A laboratory manual (2nd ed). New York: Cold Spring Harbor Laboratory Press; 1989.
- Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 1987;155:335-50.
- [Google Scholar]
- Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16:10881-90.
- [Google Scholar]
- Stabilized, freeze-dried PCR mix for detection of mycobacteria. J Clin Microbiol. 1998;36:1798-800.
- [Google Scholar]
- Amplification of ST50 gene using dry-reagent-based polymerase chain reaction for the detection of Salmonella typhi. Diagn Microbiol Infect Dis. 2007;59:373-7.
- [Google Scholar]
- Development of a dry reagent-based triplex PCR for the detection of toxigenic and non-toxigenic Vibrio cholerae. J Med Microbiol. 2011;60:481-5.
- [Google Scholar]
- Dry-reagent-based PCR as a novel tool for the rapid detection of Clostridium spp. J Med Microbiol. 2013;62:1588-91.
- [Google Scholar]
- Rapid detection of methicillin-resistant Staphylococcus aureus by a newly developed dry reagent-based polymerase chain reaction assay. J Microbiol Immunol Infect. 2014;47:484-90.
- [Google Scholar]
- Thermostabilization of indigenous multiplex polymerase chain reaction reagents for detection of enterotoxigenic Staphylococcus aureus. J Microbiol Immunol Infect. 2018;51:191-8.
- [Google Scholar]
- Development of a dry-reagent mix-based polymerase chain reaction as a novel tool for the identification of Acinetobacter species and its comparison with conventional polymerase chain reaction. J Lab Physicians. 2018;10:68-72.
- [Google Scholar]
- Dry-reagent-based PCR as a novel tool for laboratory confirmation of clinically diagnosed Mycobacterium ulcerans-associated disease in areas in the tropics where M. ulcerans is endemic. J Clin Microbiol. 2005;43:271-6.
- [Google Scholar]
- Dry reagent-based polymerase chain reaction compared with other laboratory methods available for the diagnosis of buruli ulcer disease. Clin Infect Dis. 2007;45:68-75.
- [Google Scholar]
- Ambient stable quantitative PCR reagents for the detection of Yersinia pestis. PLoS Negl Trop Dis. 2010;4:e629.
- [Google Scholar]
- Rapid detection of pathogenic leptospires by lyophilized reagent-based polymerase chain reaction. Trop Biomed. 2011;28:497-505.
- [Google Scholar]
- Development of a dry-reagent-based qPCR to facilitate the diagnosis of Mycobacterium ulcerans infection in endemic countries. PLoS Negl Trop Dis. 2015;9:e0003606.
- [Google Scholar]
- Rapid diagnosis of avian influenza virus in wild birds: Use of a portable rRT-PCR and freeze-dried reagents in the field. J Vis Exp 2011 pii: 2829
- [Google Scholar]
- Development of lyophilized loop-mediated isothermal amplification reagents for the detection of leptospira. Mil Med. 2016;181:227-31.
- [Google Scholar]
- Single-tube nested PCR with room-temperature-stable reagents. PCR Methods Appl. 1995;4:376-9.
- [Google Scholar]
- Extensive genomic diversity in pathogenic Escherichia coli and Shigella strains revealed by comparative genomic hybridization microarray. J Bacteriol. 2004;186:3911-21.
- [Google Scholar]
- Polynucleotide sequence relatedness among three groups of pathogenic Escherichia coli strains. Infect Immun. 1972;6:308-15.
- [Google Scholar]
- Molecular evolutionary relationships of enteroinvasive Escherichia coli and Shigella spp. Infect Immun. 2004;72:5080-8.
- [Google Scholar]




