
Translate this page into:
A descriptive pilot study of mitochondrial mutations & clinical phenotype in fibromyalgia syndrome
For correspondence: Dr Debashish Danda, Department of Clinical Immunology & Rheumatology, Christian Medical College & Hospital, Vellore 632 004, Tamil Nadu, India e-mail: debashisdandacmc@hotmail.com
-
Received: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
Fibromyalgia syndrome (FMS) is one of the most common chronic pain conditions of unknown aetiology. Mitochondrial dysfunction has been reported in FMS with some studies reporting the presence of mitochondrial mutation namely A3243G, which also causes mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes. This pilot study was conducted to assess this mutation and also detect large deletions in mitochondrial DNA (mtDNA) in patients with FMS.
Methods:
Thirty female patients with FMS participated and 30 matched controls were included. Genomic DNA was subjected to polymerase chain reaction (PCR) amplification using specific primers followed by restriction digestion with ApaI enzyme to detect the specific A3243G mtDNA mutation. Long-range PCR was done in two sets to detect the large deletions in the mtDNA. Biochemical parameters including thyroid-stimulating hormone and vitamin D levels were also looked at.
Results:
None of the patients were found to carry the common mutation or large deletions. Low vitamin D level was a common finding. Hypothyroidism was found in a few patients.
Interpretation & conclusions:
Although the common mutation or large mtDNA deletions were not detected in blood mtDNA in the FMS patients, mutations in the muscle and sequence variation in mtDNA remained a possibility. Future studies in both blood and muscle tissue including mtDNA sequencing are warranted in such patients to determine if a subset of FMS patients have mitochondrial myopathy.
Keywords
DNA deletions
fibromyalgia
genetic
Indian
long-range polymerase chain reaction
mitochondrial mutations
vitamin D

Fibromyalgia syndrome (FMS) is one of the most common chronic pain conditions that causes intense pain in the body, including muscles, connective tissues and joints. The disorder is estimated to affect 3-6 per cent of the world population1. While 75-90 per cent of the people who have FMS are women, it also occurs in men and children of all ethnic groups2. Diagnosis is based on the criteria given by the American College of Rheumatology (ACR)3. Fibromyalgia may be associated with psychiatric conditions such as depression and anxiety and stress-related disorders such as post-traumatic stress disorder. It leads to loss of quality life and though not life-threatening causes heavy economic burden on the society2.
The pathogenesis of FMS is not known. Psychological factors, stress and chronic sleep disturbances, brain chemical and hormonal abnormalities, mitochondrial abnormalities, polymorphisms of the serotonin genes and nuclear DNA fragmentation have been implicated4, but convincing evidence is lacking. Oxidative stress markers are also proposed as related event in its pathogenesis2. Decreased coenzyme Q10 (CoQ10) levels and increased reactive oxygen species (ROS) production in blood mononuclear cells of FMS patients are a direct evidence of increased oxidative stress at the cellular level; therefore, the therapeutic role of CoQ10 has been studied45. A study done on 30 patients and controls of FMS implied mitochondrial dysfunction as cause of inflammation6. Higher levels of serum tumour necrosis factor (TNF)-α were noted in FMS patients in this study, and increased transcript levels of TNF-α mRNA was also found in patients as compared to controls5. This suggested that FMS could be associated with immune dysregulation. Reports of mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAs) syndrome and mitochondrial myopathy have been documented in FMS78. Mutation in cytochrome b gene of mitochondrial DNA (mtDNA) in a family with FMS has been described9. With this background, we carried out clinical phenotyping and molecular studies to detect abnormalities in the germline mitochondrial gene mainly to look at the common MELAs associated point mutation, A3248G and large deletions of the mtDNA in patients with FMS.
Material & Methods
In this pilot study, 30 patients of FMS fulfilling the ACR classification of year 1990 or 2010123 or both and 30 age- and sex-matched healthy controls who were hospital staff from the same socio-cultural background were included in the study after obtaining informed written consent from 2011 to 2013. The study was done in the department of Clinical Immunology and Rheumatology in Christian Medical College, Vellore, India, and patients above 18 yr of age were selected after fulfilling the exclusion and inclusion criteria as per the ACR criteria3. Blood sample (10 ml) was taken from the patients and controls for genetic analysis. The study protocol was approved by the Institutional Review Board.
Clinical and biochemical examination: The diagnosis was based on the classification for FMS by the ACR in 1990 and/or 20103. Fibromyalgia patients must have widespread pain in all four quadrants of their body for a minimum of three months and at least 11 of the 18 specific tender points should be present. Many people who have less than 11 of the required tender points may still be diagnosed with FMS as long as they have widespread pain and many of the common symptoms associated with FMS. The suspected fibromyalgia patients underwent biochemical examination viz. creatinine, C-reactive protein (CRP), creatine phosphokinase (CPK), lactate and thyroid-stimulating hormone (TSH) whenever possible. Vitamin D levels were also evaluated in majority of the patients except in three.
Detection of common mutation: A3243G by PCR/RFLP: Whole genomic DNA was extracted from blood using Qiagen DNA extraction mini kit (Qiagen, USA). The genomic DNA was subjected to PCR. The PCR was performed using oligonucleotide primers (Sigma-Aldrich, USA): A3243G F:5’CCTCCCTGTACGAAAGGACA (3116-3135) and A3243G R:3’GCCTAGGTTGAGGTTGACCA (3609-3590) with genomic DNA of 50-100 ng/μl. The PCR product yielded a fragment of 494 bp (numbers in parentheses indicate nucleotide position on mtDNA according to Cambridge sequence)1011 on 2 per cent agarose gel electrophoresis. Amplification was performed for 35 cycles with denaturation at 95°C for 10 min, annealing at 58°C for 30 sec and extension at 72°C for 30 sec. After amplification, 10 units of Acetobacter pasteurianus sub. pasteurianus1 (Apa1) (New England Biolabs, USA) restriction enzyme was added into 10 μl of PCR products and restriction enzyme digestions were performed at 37°C for 12 h. After digestion, the products were checked on 2 per cent agarose gel electrophoresis as shown in Fig. A.
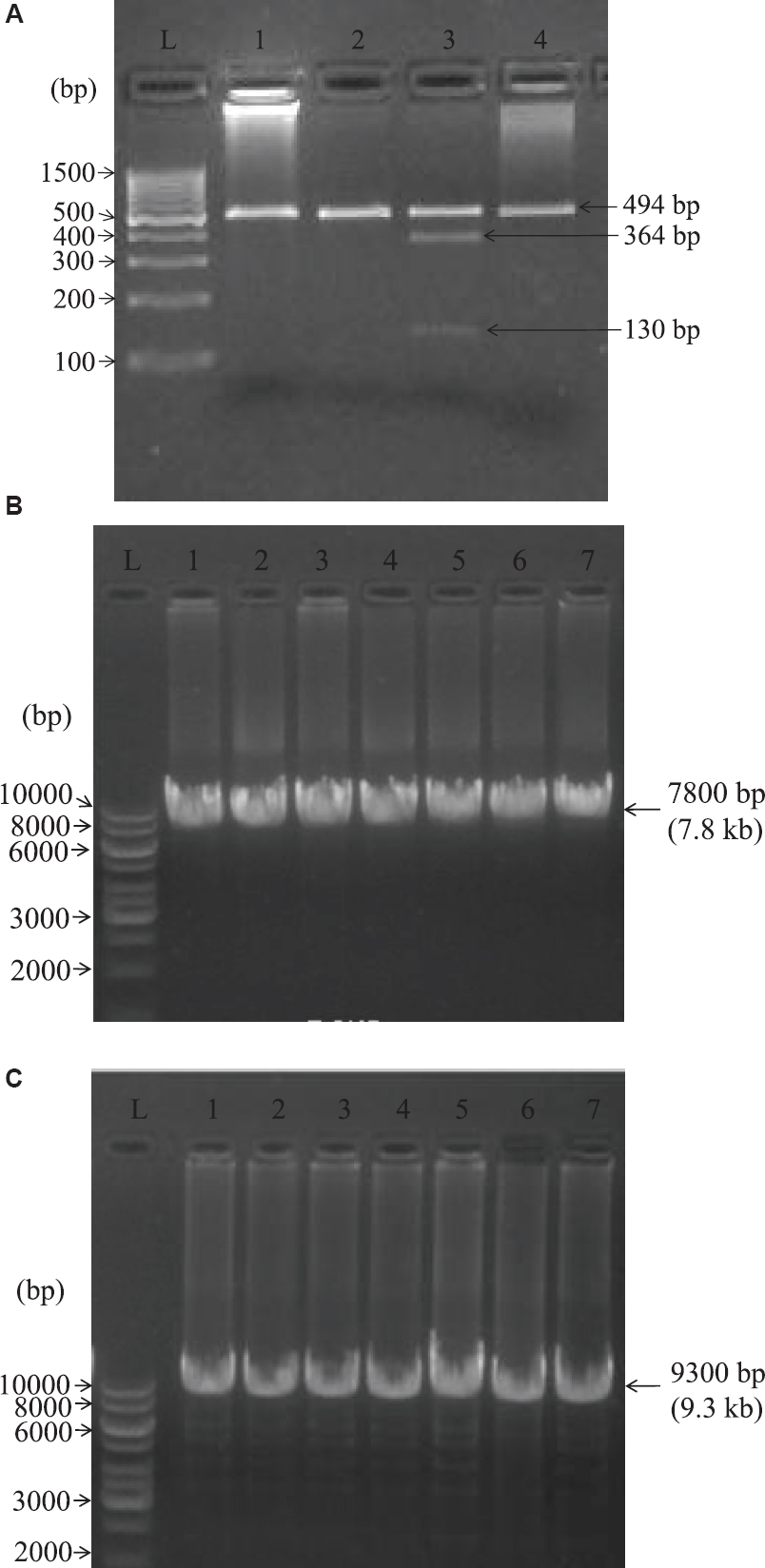
- Restriction fragment length polymorphism (RFLP) results of PCR products digested with Apa1 enzyme. (A) Lanes 1, 2 and 4 bands were negative for A3243G mutation. In lane 3, the 494 bp products with A3243G mutation were cleaved into 364 and 130 bp fragments (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes DNA-positive control). L denotes 100 bp DNA ladder. (B) Lanes 1-7 indicate the number of amplified DNA and L denotes 1 kb DNA ladder. The mitochondrial genome of all the patients was amplified in 7800 bp (7.8 kb) indicating the absence of large scale deletions. (C) Lanes 1-7 indicate the number of amplified DNA and L denotes 1 kb DNA ladder. The mitochondrial genome of all the patients was amplified in 9300 bp (9.3 kb) indicating the absence of large scale deletions.
Analysis of large-scale deletions by long-range PCR: As per the modified protocol described by Zhang et al12, the whole mtDNA (16.5 kb) was divided into two segments of 9.3 and 7.8 kb. The same genomic DNA of patients and controls were used to perform long-range PCR reaction using TaKaRa LA PCR kit ver.2.1 (Takara Bio Inc., USA) along with two segments of oligonucleotide primers (Sigma); amplification was performed for 9.3 kb segment for 30 cycles with F:5’ACATAGCACATTACA GTCAAATCCCTTCTCGTCCC and R:3’ATTG CTAGGGTGGCGCTTCCAATTAGGTGC under the following thermocycling conditions: denaturation at 94°C for 30 sec, annealing at 68°C for nine min and 68°C extension for 13 min. The PCR products were loaded on 1 per cent agarose gel in 1× tris acetate EDTA (TAE) buffer along with 1 kb ladder for 45 min - 1 h at 120 volts (Fig. B). The same genomic DNAs of patients and controls were used to perform long-range PCR using 7.8 kb oligonucleotide primers: F:5’TCATTTTTATTGCCACAACTAAC CTCCTCGGACTC and R:3’CGTGATGTCTTAT TTAAGGGGAACGTGTGGGCTAT. The reactions were performed using TaKaRa LA PCR kit ver 2.1 for 30 cycles under the following thermocycling conditions: denaturation at 94°C for 30 sec, annealing at 68°C for eight min and extension at 68°C for 13 min. The post-PCR products were checked on 1 per cent agarose gel in 1× TAE buffer for 45 min - 1 h at 120 volts (Fig. C).
Results
The age of patients ranged from 27 to 67 yr with a mean of 49 yr. Serum CPK was normal in all except in one patient who had a marginal elevation of 201 (U/l) normal range being up to 200. Otherwise, the average value was 97 with range of 47-201 U/l. Serum lactate levels were normal in all with an average of 1.25 mmol/l. Vitamin D levels done in 27 out of 30 were in the deficiency range (20 ng/dl) in 17 patients, while nine patients had normal vitamin D (21-29 ng/dl); only one patient had value above 30 ng/dl. Serum creatinine, CRP, serum glutamic pyruvic transaminase and TSH levels were in normal range. Three patients had elevated TSH of whom one was started on small dose of hormone replacement, one was lost to follow up and another had a repeat TSH in the normal range.
Detection of A3243G mutation: As shown in the Fig. A, the wild mtDNA showed a band of 494 bp after Apa1 digestion, which indicated the absence of A3243G mutation in fibromyalgia patients sample. In the mutant mtDNA taken as positive control from a proven MELAS patient harboring the A3243G mutation, the 494 bp product was cleaved into 364 and 130 bp showing two bands. Large-scale deletions of the mitochondrial genome of all the patients were amplified in 7800 and 9300 bp which indicated absence of large scale deletions (Fig. B and C).
Discussion
This pilot study of 30 patients with FMS was done to rule out germline mitochondrial mutations or deletions as a possible aetiological factor in a subset of FMS patients. None of the patients had biochemical parameters suggestive of mitochondrial cytopathy except for one who had marginal elevation of serum CPK. The common mutation A3243G and large deletions were not detected in germline mtDNA in these patients including the one with elevated CPK. DNA fragmentation and ultrastructural changes in mitochondria were detected by Sprott et al4. Occasional cases reports of FMS presenting with MELAS were found to have the common mutation when tested78. Report of Cordero et al9 detecting mutation in cytochrome b gene of mtDNA supports need of further studies. Mitochondrial dysfunction related inflammation has been implicated in FMS and CoQ10 was given for therapeutic benefit1314. This study14 showed the beneficial effect of CoQ10 in reducing the symptoms of FMS and chronic fatigue syndrome a condition akin to FMS. Expression of mitochondrial disorders is also complicated by homoplasmy and heteroplasmy in different tissues and hence it may be difficult to detect germline mutation. However, muscle biopsy was not done in our patients with FMS.
Bhatty et al15 and our study found low vitamin D levels in majority of the patients. It is possible that abnormalities in TSH and CPK and low vitamin D levels noted in our FMS patients is a reflection of the abnormalities seen in general population. However, these parameters are treatable and can be often corrected to relieve the patients of symptoms and hence require assessment if missed in earlier evaluation of FMS patients.
Occasional case reports have suggested that a subset of FMS patients may have mitochondrial disease; however, the actual prevalence of this in true FMS cases is not known. Our study did not show the common mutation or large scale deletions. In future, larger studies along with mitochondrial gene sequencing are needed to address this question.
Financial support & sponsorship: Funding was provided by Lady Tata foundation for Research, Mumbai.
Conflicts of Interest: None.
References
- Is inflammation a mitochondrial dysfunction-dependent event in fibromyalgia? Antioxid Redox Signal. 2013;18:800-7.
- [Google Scholar]
- Effects of vitamin D therapy on quality of life in patients with fibromyalgia. Eurasian J Med. 2017;49:113-7.
- [Google Scholar]
- The American college of rheumatology preliminary diagnostic criteria for fibromyalgia and measurement of symptom severity. Arthritis Care Res (Hoboken). 2010;62:600-10.
- [Google Scholar]
- Increased DNA fragmentation and ultrastructural changes in fibromyalgic muscle fibres. Ann Rheum Dis. 2004;63:245-51.
- [Google Scholar]
- Oxidative stress in fibromyalgia: Pathophysiology and clinical implications. Reumatol Clin. 2011;7:281-3.
- [Google Scholar]
- Mitochondrial dysfunction and mitophagy activation in blood mononuclear cells of fibromyalgia patients: Implications in the pathogenesis of the disease. Arthritis Res Ther. 2010;12:R17.
- [Google Scholar]
- Mitochondrial encephalomyopathy with lactic acidosis and strokelike episodes (MELAS): A mitochondrial disorder presents as fibromyalgia. South Med J. 2004;97:528-31.
- [Google Scholar]
- Mitochondrial myopathy presenting as fibromyalgia: A case report. J Med Case Rep. 2012;6:55.
- [Google Scholar]
- Mutation in cytochrome b gene of mitochondrial DNA in a family with fibromyalgia is associated with NLRP3-inflammasome activation. J Med Genet. 2016;53:113-22.
- [Google Scholar]
- Mitochondrial gene mutations in the tRNA(Leu(UUR)) region and diabetes: Prevalence and clinical phenotypes in Japan. Clin Chem. 2001;47:1641-8.
- [Google Scholar]
- The case for the continuing use of the revised Cambridge Reference Sequence (rCRS) and the standardization of notation in human mitochondrial DNA studies. J Hum Genet. 2014;59:66-77.
- [Google Scholar]
- Comprehensive one-step molecular analyses of mitochondrial genome by massively parallel sequencing. Clin Chem. 2012;58:1322-31.
- [Google Scholar]
- Coenzyme Q(10): A novel therapeutic approach for fibromyalgia? Case series with 5 patients. Mitochondrion. 2011;11:623-5.
- [Google Scholar]
- Does oral coenzyme Q10 plus NADH supplementation improve fatigue and biochemical parameters in chronic fatigue syndrome? Antioxid Redox Signal. 2015;22:679-85.
- [Google Scholar]




