
Translate this page into:
Identification of three novel mutations in SLCO2A1 in Asian-Indians with Pachydermoperiostosis
*For correspondence: shagun.genetics@gmail.com
-
Received: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Sir,
Pachydermoperiostosis (PDP) or primary hypertrophic osteoarthropathy, is a rare genetic disorder characterized by pachydermia (thickening of the skin), clubbing (acropathy) and periostitis with new bone formation1. It is inherited in both autosomal dominant (OMIM#167100) and autosomal recessive (OMIM #259100, OMIM#614441) forms. In 1935, Touraine, a French dermatologist, classified it into three clinical types namely complete (periostitis, pachyderma including cutis verticis gyrata), incomplete (without pachyderma) and forme fruste (pachyderma with minimal skeletal changes)2. The genetic basis of PDP was first determined by the identification of homozygous variants in 15-hydroxyprostaglandin dehydrogenase (HPGD), the chief enzyme involved in prostaglandin degradation3. A second causative gene, SLCO2A1, which encodes for prostaglandin (PGE2) transporter mediates the uptake of PGE2 across the plasma membrane, was subsequently implicated in both autosomal recessive as well as dominant forms of the disease4. Variants in these two genes lead to the elevation of extracellular PGE2 levels, which result in the constellation of symptoms. So far, 50 different disease-causing variants in SCLO2A1 have been reported, while HPGD gene variants are rarer, with 14 different variants having been reported5. We report three cases of PDP from unrelated families of Asian-Indian ethnicity, associated with variants in the SCLO2A1 gene.
Three individuals presenting to two different insititutes are discussed here. An informed consent was sought from each of them. Cases 1 and 2 were recruited from Nizam’s Institute of Medical Sciences, Hyderabad and case 3 was recruited from Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow. Molecular characterization and reporting were done between 2019 and 2021, at the Centre for DNA Fingerprinting and Diagnostics (CDFD), Hyderabad, after approval from the Institutional Ethics Committee of CDFD.
Case 1: A 25 yr old proband of south Indian ethnicity, born to a healthy third degree consanguineous parents, presented on December 26, 2013, with enlarged hands and feet, and swelling and pain in knee joint which gradually worsened over the prior three years. He was diagnosed with juvenile arthritis and treated symptomatically. There was no family history of similar complaints. On evaluation, coarse facial skin with marked cutis gyrata (grade 2 pachyderma), heavy eyelids with ptosis, blepharoptosis and seborrhoea with oily facial skin was appreciated. Clubbing of hands and feet was prominent. Significant swelling over the knee and ankle joints was present without a local rise in temperature, tenderness or limitation of joint mobility (Fig. 1A-C). Skeletal survey showed cortical thickening and sclerosis of the tubular bones with marked periosteal reaction, most notably at the distal portions of the radii, ulnae, tibiae and fibulae. There was mild expansion of the diaphysis of the long tubular bones. Diagnosis of PDP was made clinically as he had all the pathognomonic clinical features.
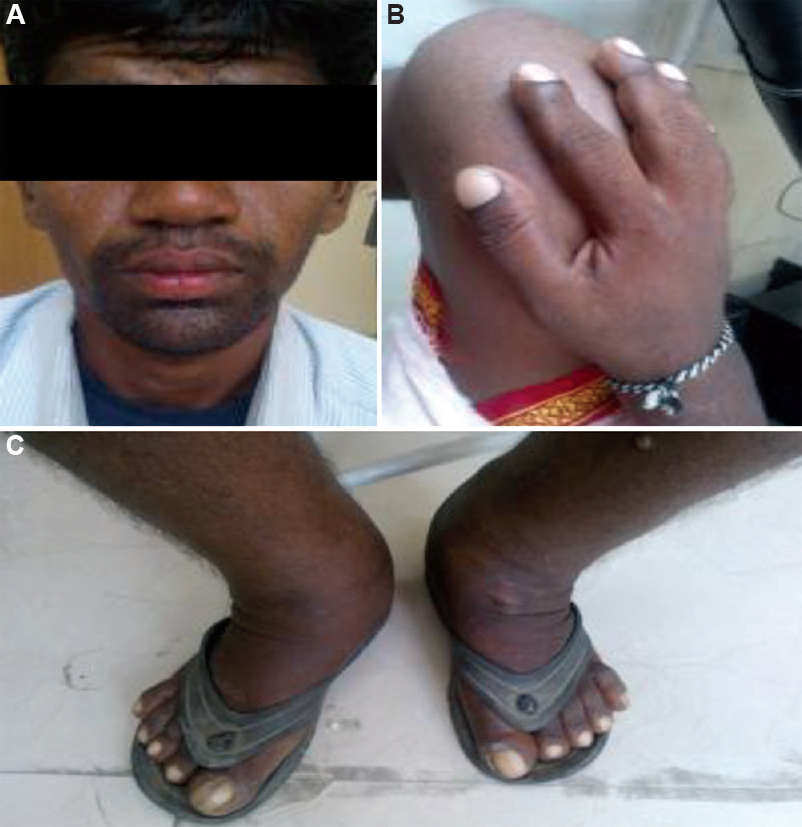
- (A) Coarse facial skin with marked cutis gyrate (grade 2 pachyderma), heavy eyelids with ptosis, blepharoptosis and seborrhoea with oily facial skin. (B) Clubbing of hand. (C) Significant swelling over legs and ankle joints with clubbing.
Case 2: A 23 yr old male born to a third degree consanguineous couple of south Indian ethnicity, was referred from the orthopaedic department and presented on July 20, 2018, with complaints of pain and swelling of bilateral ankle joints, excessive sweating and recurrent intermittent fever for the past six years. A history of intermittent joint stiffness was present in the large proximal joints. Family history of similar complaints was present in the paternal uncle. Physical examination revealed enlargement of the terminal phalanges and facial furrowing with grade 1 to 2 pachyderma (cutaneous thickening with mild puckering). There was bilateral ankle swelling, but knee joints were normal (Fig. 2A-C). Skeletal survey showed periosteal thickening of the long bones with marked periosteal reaction and areas of diffuse sclerosis along with coarse trabeculations (Fig. 2D-F). He showed a milder disease than case 1.
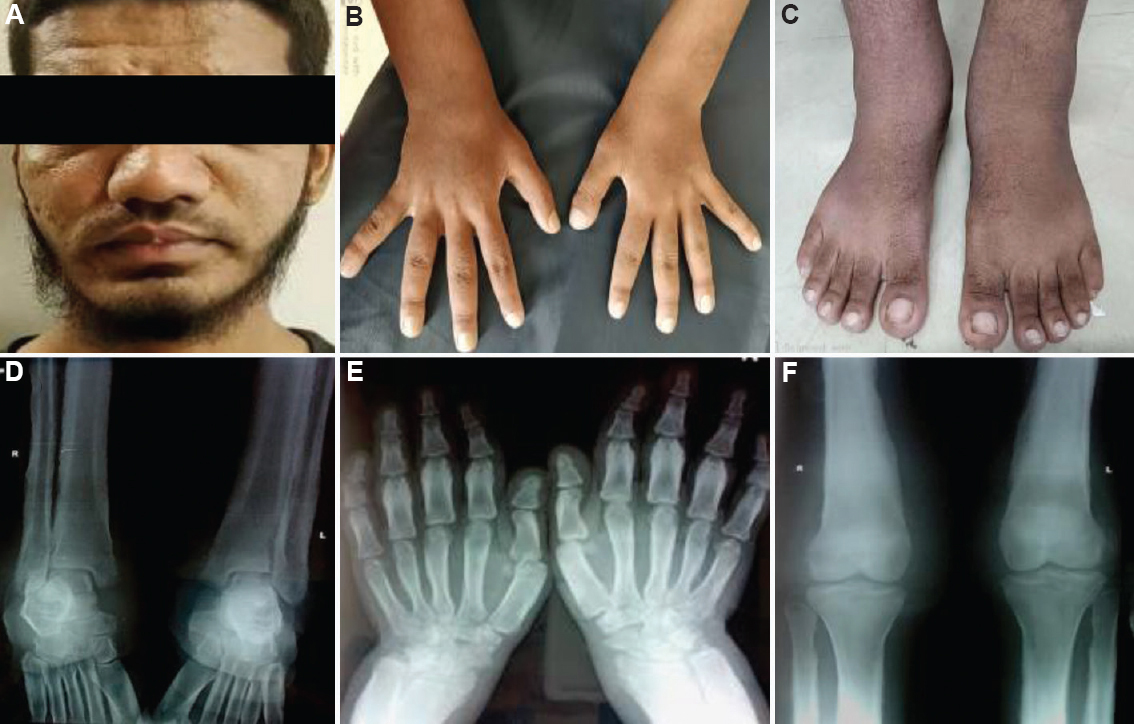
- (A) Grade 1 to 2 pachyderma with excessive sweating with facial furrowing. (B) Clubbing of hands. (C) Ankle joint swelling. (D) Diaphyseal expansion, periosteal reaction and rarefaction of spongiosa with coarse trabeculation in the distal end of tibiae and fibulae. (E) Diffuse sclerosis seen in the lower end of radius and ulna with cortical sclerosis. (F) Cortical thickening, sclerosis of the tubular bones with the marked periosteal reaction of the distal end of the femur and proximal tibia.
Case 3: A 20 yr old male, born to a non-consanguineous couple of north Indian ethnicity, presented on December 22, 2014, with bilateral swelling of ankle joints. On examination, he had grade 1 pachyderma with thickening of facial skin without facial puckering, oedema of bilateral ankle joints with grade 5 clubbing was seen (Fig. 3D-F). Skeletal survey showed cortical thickening, diaphyseal widening and periosteal reaction along shafts of long bones (Fig. 3A-C). Overall, these case had a milder phenotype as compared to 1 and 2.
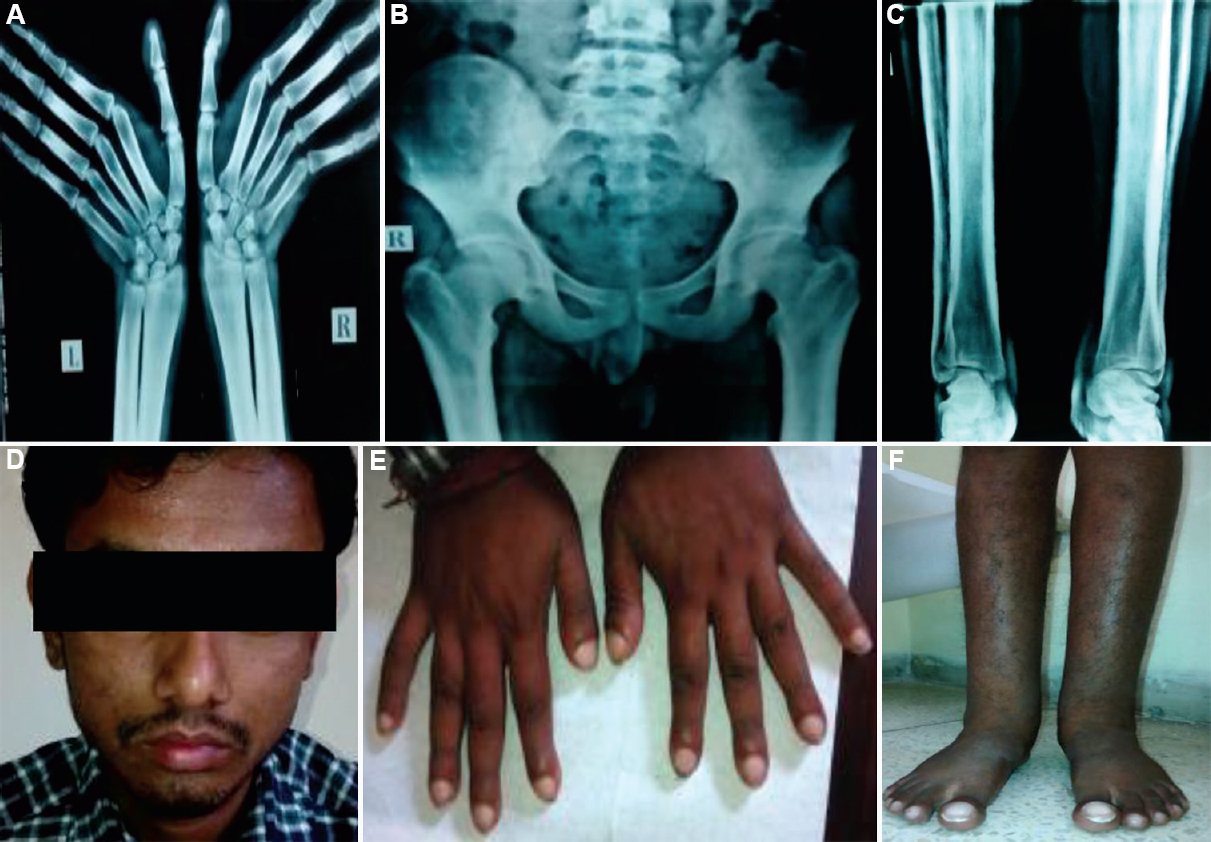
- (A) Diffuse sclerosis and the periosteal reaction seen in the lower end of radius and ulna with the transverse sclerotic band. (B) Sclerotic bands along the inferior margin of iliac bones with coarse trabeculation of the iliac wings. (C) Cortical thickening, diaphyseal widening and periosteal reaction along shafts of long bones of legs. (D) Grade 1 pachyderma with thickening of facial skin without facial puckering. (E) widening of tips of fingers with grade 5 clubbing. (F) Bilateral leg and ankle joint swelling with clubbing.
Molecular characterization of HPGD and SLCO2A1: DNA extraction from peripheral blood leucocytes was done, followed by PCR and Sanger sequencing of the exonic as well as the flanking intronic regions of the HPGD and SLCO2A1 genes. Variants underwent in silico characterization using ClustalW sequence alignment and a study of the impact of this missense variant on protein sequence and structure (Supplementary Methods).
Our analysis revealed one missense, one nonsense and one deletion variant in SLCO2A1 in the three cases, respectively (Supplementary Table I). In cases 1, a homozygous novel missense variant c.614C>T (p.Pro205Leu) in exon 4 was identified. This was predicted to be disease causing by Mutation Taster (https://www.mutationtaster.org/), PolyPhe-2 and the amino acid shows evolutionary conservation (
| Participants | Age (yr) | Variant | Protein variant | Location | Gene | Type of mutation | Zygosity |
|---|---|---|---|---|---|---|---|
| Case 1 | 25 | c.614C>T | p.P205L | Exon 4 | SLCO2A1 | Missense | Homozygous |
| Case 2 | 23 | c.529C>T | p.Q177* | Exon 4 | SLCO2A1 | Nonsense | Homozygous |
| Case 3 | 20 | c.1237_1246delTCCTTTGTGT | V413Sfs*64 | Exon 9 | SLCO2A1 | Frameshift | Homozygous |
| Participants | Nucleotide pathogenic variation [(allele 1)+(allele 2)] | Protein pathogenic variation [(allele 1)+(allele 2)] | Known/novel | Mutation taster | SIFT | Polyphen | Clinvar submission ID |
|---|---|---|---|---|---|---|---|
| Case 1 | c.614C>T/c.614C>T | p.P205L/p.P205L | Novel | Disease-causing | Affect protein function | Probably damaging | SUB9840934 |
| Case 2 | c.529C>T/c.529C>T | p.Q177*/p.Q177* | Novel | Disease-causing | NA | NA | SUB9840934 |
| Case 3 | c.1237_1246delTCCTTTGTGT/c. 1237_1246delTCCTTTGTGT | V413Sfs*64/V413Sfs*64 | Novel | Disease-causing | NA | NA | SUB9840934 |
SIFT, sorting intolerant from tolerant; NA, not available
In addition to other clinical presentations, such as hyperhidrosis, arthralgia, gastric ulcers, seborrhoea, acne and joint effusions, occasionally, congenital cardiac disease (patent ductus arteriosus), myelofibrosis, enteropathy, acromegaly and soft-tissue tumours are also reported in PDP5. Elevated PGE2 levels resulting from reduced degradation (HPGD associated) or abnormal transport across the plasma membrane (SCLO2A1 associated) are implicated in the pathogenesis of this disease6. PGE2 induces cytokine-mediated tissue remodelling and vascular stimulation, causing hyperhidrosis, acro-osteolysis, periostitis, pachyderma and arthritis7. Previously, four patients of Asian-Indian ancestry presenting with a mild form of PDP have been reported, further reflecting the clinical variability of this disorder8.
As reportedly previously, males were more commonly and severely affected, with the male:female ratio being 9:17. A few females carrying the SLCO2A1 mutation have been reported to have presented with mild or atypical symptoms7. In this study, all probands were male. Although no definite biological studies were performed, it has been hypothesized that the difference in the interaction of testosterone and estrogen on the mediators of the COX-2 pathway plays a role in the skewed male:female ratio7.
Although PDP is widely regarded as autosomal recessive disorder, autosomal dominant transmission with incomplete penetrance has been reported previously9 which is in line with the findings of the present study, where all patients showed an autosomal recessive inheritance.
So far, 50 different SLCO2A1 mutations have been reported10. This study revealed three novel variants in patients of Asian-Indian ethnicity. Previously, a study by Radhakrishnan et al11. reported four patients of Asian-Indian ethnicity with variants in HPGD11. Busch et al12 have previously reported two brothers with Asian-Indian ethnicity with a homozygous missense mutation in exon 6 of SLCO2A1 gene.
Genotype/phenotype correlation between SCLO2A1 and HPGD-associated PDP has been attempted previously. According to a study by Seta et al13, individuals with SLCO2A1 mutations tends to develop later onset, more severe disease with a greater likelihood of cutis verticis gyrata and joint involvement compared to patients with HPGD mutations. Hereditary enteropathy and myelofibrosis are also more commonly associated with SLCO2A1 mutations. Sasaki et al14 reported five individuals with SLCO2A1 variants and noted that individuals homozygous for the founder mutation c.940+1G>A had a more severe phenotype as compared to compound heterozygotes14. In the present study, case 1 who harboured the missense variant, were more severely affected compared to cases 2 and 3, who were homozygous for more deleterious alleles. Larger studies are hence needed to understand the biological basis of this and ascertain accurate genotype-phenotype correlations.
PDP can mimic common non-genetic conditions such as acromegaly, lepromatous leprosy, mucinosis, myxoedema and juvenile periostitis (syphilitic periostitis, psoriatic onycho-pachydermo periostitis and Paget’s disease)15. However, digital clubbing and periostitis are not commonly associated with acromegaly and periostitis conditions rarely have cutaneous manifestations. Further endocrine evaluations of all the three cases were normal, ruling out acromegaly and myxoedema. Sensory system examination in all three cases was normal, which is often impaired in individuals with lepromatous leprosy. Skin biopsy would have contributed significantly to ruling out mucinosis and other clinical mimickers. However, we were unable to obtain consent for skin biopsy. Furthermore, parental and additional family member testing could not be done in two of the families due to sample unavailability. Case 2 had a paternal uncle with similar manifestations, who could not be evaluated.
Incomplete forms of PDP can pose a diagnostic challenge and systemic examination, endocrine evaluation and skin biopsy can be of great help in ruling out the clinical mimics. Elevated urinary excretion of PGE2 and PGE-M in patients with PDP can be helpful in early and differential diagnosis of PDP, assessment of severity and monitoring of disease. Therapeutic options include salicylates, non-steroidal anti-inflammatory drugs, systemic corticosteroids, hydroxyurea and colchicine. Bisphosphonates or infliximab have been attempted in cases of PDP refractory to conventional therapy16. Cosmetic surgery or botulinum toxin injections can be useful for correcting rough facial features.
Overall, mutations in the SCLO2A1 gene are commonly involved in the aetiology of PDP is in Asian-Indian patients. All three variants described in this study are novel variants contributing to the mutation spectrum of this disease.
Financial support and sponsorship
This work is supported by the in-house research funds (Grant No. BTl/AAQ/01/CDFD-Flagship/2019) of the Centre for DNA Fingerprinting and Diagnostics, Hyderabad.
Conflicts of interest
None.
Supplementary Fig. 1
Supplementary Fig. 1 (A) Human SLCO2A1 gene amino acid sequence of p.Pro205Leu showing highly evolutionary conservation with other species, (B) Multiple sequence alignment using ClustalW. Structural analysis of SLCO2A1 showing variation in polar contacts in the wild type (WT) (left panels), and in corresponding mutant Pro205Leu (Right panel).Supplementary Fig. 2
Supplementary Fig. 2 Exon-wise representation of the three different disease-causing novel variants in SLCO2A1.The exonic number is mentioned on the corresponding exon box.Supplementary Fig. 3
Supplementary Fig. 3 Sanger sequence chromatograms of SLCO2A1 gene in three patients and available parents.References
- Complete primary pachydermoperiostosis:A case report from Jordan and review of literature. Clin Case Rep. 2019;7:346-52.
- [Google Scholar]
- Un syndrome osteodermopathique:La pachydermie plicaturee avec pachyperiostose ds extremites. Press Med. 1935;43:1820-4.
- [Google Scholar]
- Mutations in 15-hydroxyprostaglandin dehydrogenase cause primary hypertrophic osteoarthropathy. Nat Genet. 2008;40:789-93.
- [Google Scholar]
- Exome sequencing identifies SLCO2A1 mutations as a cause of primary hypertrophic osteoarthropathy. Am J Hum Genet. 2012;90:125-32.
- [Google Scholar]
- A common mutation and a novel mutation in the HPGD gene in nine patients with primary hypertrophic osteoarthropathy. Calcif Tissue Int. 2015;97:336-42.
- [Google Scholar]
- Mutations in the prostaglandin transporter encoding gene SLCO2A1 cause primary hypertrophic osteoarthropathy and isolated digital clubbing. Hum Mutat. 2012;33:660-4.
- [Google Scholar]
- Identification of the mutations in the prostaglandin transporter gene, SLCO2A1 and clinical characterization in Korean patients with pachydermoperiostosis. J Korean Med Sci. 2016;31:735-42.
- [Google Scholar]
- Milder form of pachydermoperiostosis:A report of four cases. Clin Dysmorphol. 2009;18:85-9.
- [Google Scholar]
- Monoallelic mutations in SLCO2A1 cause autosomal dominant primary hypertrophic osteoarthropathy. J Bone Miner Res. 2021;36:1459-68.
- [Google Scholar]
- Identification of mutations in the prostaglandin transporter gene SLCO2A1 and phenotypic comparison between two subtypes of primary hypertrophic osteoarthropathy (PHO):A single-center study. Bone. 2018;106:96-102.
- [Google Scholar]
- Digital clubbing as the predominant manifestation of hypertrophic osteoarthropathy caused by pathogenic variants in HPGD in three Indian families. Clin Dysmorphol. 2020;29:123-6.
- [Google Scholar]
- Mutations in the prostaglandin transporter SLCO2A1 cause primary hypertrophic osteoarthropathy with digital clubbing. J Invest Dermatol. 2012;132:2473-6.
- [Google Scholar]
- Pachydermoperiostosis:The value of molecular diagnosis. Ann Dermatol Venereol. 2017;144:799-803.
- [Google Scholar]
- Identification of mutations in the prostaglandin transporter gene SLCO2A1 and its phenotype-genotype correlation in Japanese patients with pachydermoperiostosis. J Dermatol Sci. 2012;68:36-44.
- [Google Scholar]
- Pachydermoperiostosis mimicking acromegaly:A case report. Indian Dermatol Online J. 2018;9:182-4.
- [Google Scholar]
- Bisphosphonates use in pachydermoperiostosis. J Assoc Physicians India. 2019;67:87-90.
- [Google Scholar]
Supplementary Methods
Isolation of DNA and PCR: Genomic DNA was isolated from the peripheral white blood cell from patients and their parents using DNA extraction and purification kit (Qiagen, Hilden, Germany) to identify any mutations in HPGD and SLCO2A1.DNA sequences were downloaded from Ensembl Genome Browser. Primers were designed for all the exons of HPGD and SLCO2A1, including the flanking intronic regions by Primer3 Input Version 0.4.0 (frodo.wi.mit.edu/). Polymerase chain reaction (PCR) was carried out in the Bio-Rad thermocycler machine for all exons of HPGD and SLCO2A1 for under the following conditions. Patient DNA samples were amplified in 15 μl reaction using 6 μl of 2XMasterMix (Qiagen, Hilden, Germany), 100 ng/μl template (1 μl), 10 pm/μl of forward primer (1 μl), 10 pm/μl of reverse primer (1μl) buffer 3 μl and nuclease free water 3 μl. Thermocycler conditions were as follows: Intial denaturation at 95°C for 5 min, 35 cycles of denaturation at 94°C for 30 sec annealing temperature at 60°C 30 sec, extension at 72°C 1 min and final extension at 72°C for 10 min.
Sanger sequencing: All PCR products were purified according to standardized kit protocol (QIAquick Gel Extraction Kit, Qiagen, Hilden, Germany) and DNA sequencing was carried out by using capillary electrophoresis on ABI 3130 automated genetic analyzer according to the manufacturer’s instruction (Applied Biosystems, Foster City, CA). Genomic DNA sequencing data of HPGD and SLCO2A1 were analyzed using the tool EMBOSS to find sequence variations in these genes.
In silico characterization: Novel pathogenic variant prediction softwares, such as MutationTaster (http://www.mutationtaster.org/), MutationAssessor (www.mutationassessor.org), HANSA (www.hansa@cdfd.org.in), Polyphen2 (genetics.bwh.harvard.edu/pph2/) and SIFT (sift.jcvi.org), were used for In silico characterization of identified variants. The variants were also checked in databases such as dbSNP, 1000 Genome browser and gNOMAD, as well as in-house exome database for the exclusion of polymorphic variants. Variants were also checked in ClinVar and HGMD databases for known pathogenic variants in HPGD and SLCO2A1 genes. HGVS mutation nomenclature was used for reporting sequence variations (http://www.hgvs.org). Evolutionary conservation of the mutated amino acid residues was checked across different species. Segregation analysis was performed for the novel sequence variants.
Characterization and impact of novel missense variant on protein sequence and structure: In silico characterization was done for novel pathogenic variants in SLCO2A1 protein. No templates were found for SLCO2A1. We used I-TASSER (Iterative Threading ASSEmbly Refifinement) to generate a 3D-structure of wildtype. Best C-score predicted structures were downloaded from I-TASSER. The mutant structure was generated using the PyMOL mutate function. Both wild-type and mutated structures were visualized using the PyMOL tool.




