
Translate this page into:
Bioconjugate-loaded solid lipid nanoparticles for enhanced anticancer drug delivery to brain cancer cells: An in vitro evaluation
For correspondence: Dr Vandana Soni, Department of Pharmaceutical Sciences, Dr Hari Singh Gour Central University, Sagar 470 003, Madhya Pradesh, India e-mail: drvandanasoni@gmail.com
-
Received: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
The treatment of brain cancer is still challenging for an oncologist due to the presence of the blood-brain barrier (BBB) which inhibits the entry of more than 98 per cent of drugs used during the treatment of brain disease. The cytotoxic drugs used in chemotherapy for brain cancer treatment also affect the normal cells due to lack of targeting. Therefore, the objective of the study was to develop tween 80-coated solid lipid nanoparticles (SLNs) loaded with folic acid-doxorubicin (FAD) conjugate for site-specific drug delivery to brain cancer cells.
Methods:
The FAD conjugate was synthesized by the conjugation of folic acid with doxorubicin and characterized by Fourier transform infrared spectroscopy and proton nuclear magnetic resonance spectroscopy. SLNs loaded with FAD were prepared by the solvent injection method. The SLNs were characterized by the particle size, zeta potential, surface morphology, entrapment efficiency, etc.
Results:
The average particle size of FAD conjugate-loaded SLNs (SLN-C) was found to be 220.4±2.2 nm, with 36.2±0.6 per cent entrapment efficiency. The cytotoxicity and cellular uptake were determined on U87 MG cell lines. Half maximal inhibitory concentration value of the SLN-C was found to be 2.5 µg/ml, which confirmed the high antitumour activity against brain cancer cells.
Interpretation & conclusions:
The cell line studies confirmed the cytotoxicity and internalization of SLN-C in U87 MG brain cancer cells. The results confirmed that tween 80-coated SLNs have the potential to deliver the doxorubicin selectively in the brain cancer cells.
Keywords
Brain cancer
conjugate
doxorubicin
folic acid
solid lipid nanoparticles

The primary brain tumour is one of the ten main causes of death by cancer1. At present, the available treatment for brain tumour is surgery followed by radiotherapy and chemotherapy2. In spite of these available treatments, the survival rate of the brain cancer patients is low because of the presence of the blood-brain barrier (BBB)3 and blood-brain-tumour barrier (BBTB)4. BBB consists of the tight endothelial cells, which inhibit the entry of drug molecules into the brain, resulting in failure or less effective chemotherapy5-7. The BBTB also limits the accumulation of drugs inside the brain tumour cells. These limitations create obstacles during brain cancer treatment. Therefore, there is a need to develop an effective nanocarrier system which overcomes the BBB and maximize drug availability to cancer cells, with less or no accumulation of drugs in normal cells. The coating of the nanocarriers with hydrophilic surfactants has been proved as a promising approach to cross the BBB. Among the number of surfactants, tween 80 (non-ionic surfactant) proved as the most efficient surfactant to enhance the permeability of the nanocarriers across the BBB8. Different mechanisms have been proposed for the transportation of the non-ionic surfactant-coated nanocarriers, i.e. endocytosis, membrane fluidization and inhibition of P-gp efflux9-11. Hence, the objective of this study was to develop a nanocarrier system capable of delivering the active biomoeity across the BBB. To achieve the said objective, tween 80-coated solid lipid nanoparticles (SLNs) were developed which possess the advantages such as biodegradable and stable and can be taken up readily by the brain compared to other carriers12-14. The complete hypothesis is shown in Figure 1.
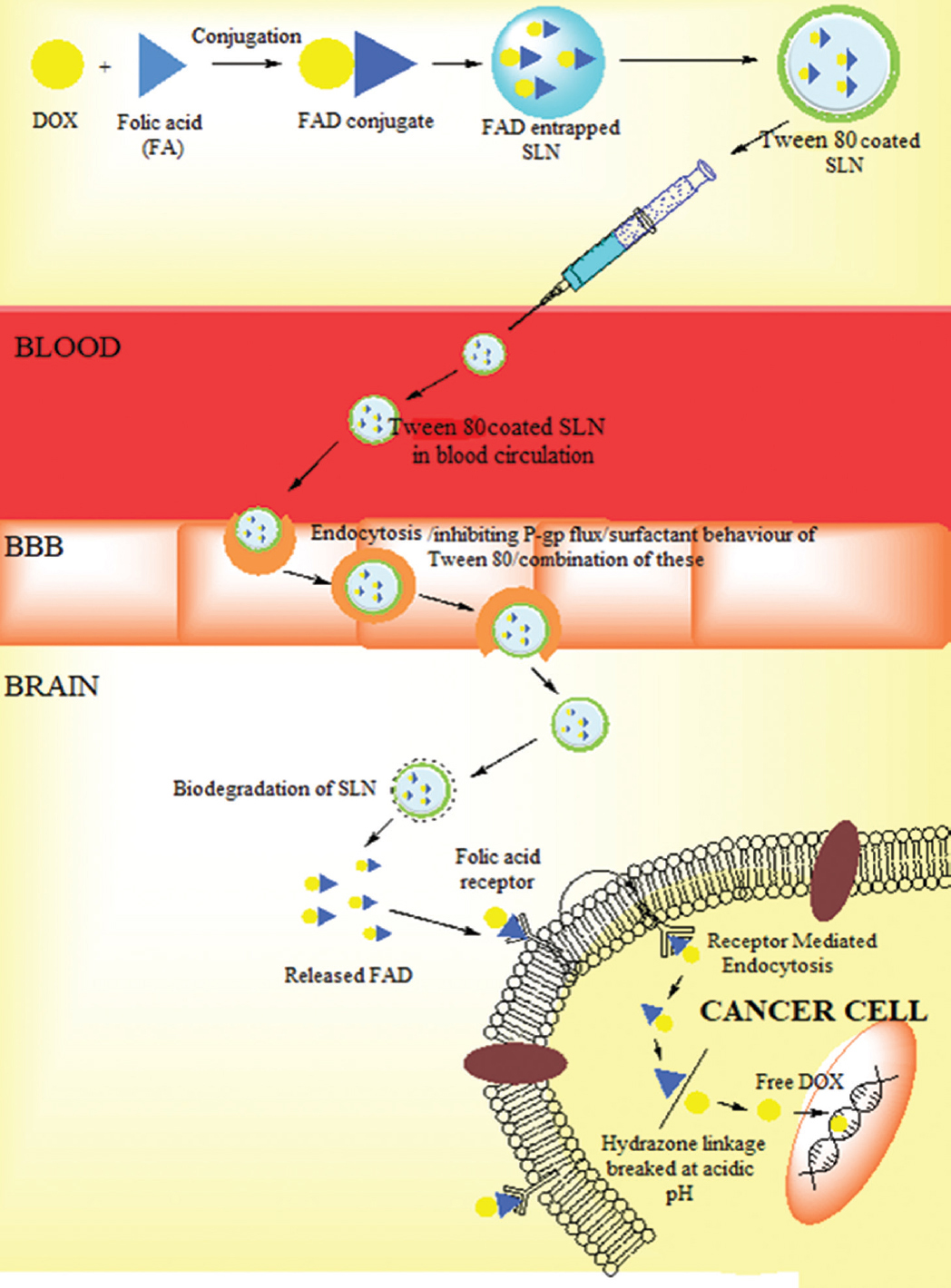
- Representation of dual targeting of solid lipid nanoparticles.
Material & Methods
Synthesis of the conjugate folic acid-aminocaproic acid-doxorubicin (FAD): The FAD was synthesized by the method reported by Ye et al15, 2013 (
Synthesis of aminocaproic acid (AMA) acetate: The AMA acetate was synthesized by addition of thionyl chloride (25 ml, Sigma-Aldrich, Mumbai, India) in ethanol (150 ml, Himedia, Mumbai) taken in a round bottom flask kept over an ice bath. The reaction was continued for 12 h with stirring. Then, AMA (0.99 mmol, Spectrochem, Mumbai) was added and again stirred for 6 h. The thionyl chloride and ethanol were removed by a rotary evaporator. The obtained powder was recrystallized by a mixture of methanol-petroleum ether [5:1 (v/v), Himedia].
Synthesis of folic acid-aminocaproic acid (FA-AMA) acetate (FAA): FA-AMA acetate (FAA) was synthesized by dissolving FA (1.99 mmol) in dimethyl sulfoxide [(DMSO) 15 ml, Himedia], NHS (2.99 mmol, Spectrochem), DCC (2.99 mmol, Spectrochem) and triethylamine [(TEA) 120 µl, Spectrochem] were added with the above reaction mixture. The reaction was maintained at room temperature (RT) with stirring for 8 h followed by the addition of AMA acetate (5.45 mmol). Further stirring for additional 12 h was continued followed by filtration. To the filtered product acetone:diethylether [1:1 (v/v), Himedia], mixture was added and the precipitate was obtained which was further filtered and washed by using diethyl ether and purified by column chromatography.
Synthesis of folic acid-aminocaproic acid (FA-AMA) hydrazide (FAH): FA-AMA hydrazide (FAH) was synthesized by the addition of hydrazine hydrate (4 ml, Spectrochem) to a solution of FAA (0.685 mmol) dissolved in DMSO. The reaction proceeded for eight hours with stirring at 50°C followed by a cooling at room temperature. The reaction was precipitated with ethanol and the precipitate filtered, washed with diethyl ether and purified by column chromatography.
Synthesis of FAD: FAD was synthesized by the addition of DOX (0.137 mmol) and trifluoroacetic acid (60 µl, Spectrochem) in a solution of FAH (0.404 mmol) in 10 ml DMSO. The reaction was continued for 12 h at RT followed by the addition of acetone-diethyl ether (1:1 v/v). The resultant precipitate was filtered, washed with diethyl ether and purified by column chromatography. The precipitate was characterized by Fourier transform infrared spectroscopy (FTIR, Shimadzu, Japan) and proton nuclear magnetic resonance spectroscopy (1H NMR spectroscopy, Bruker ACF300, Germany) (Figs 2 and 3).
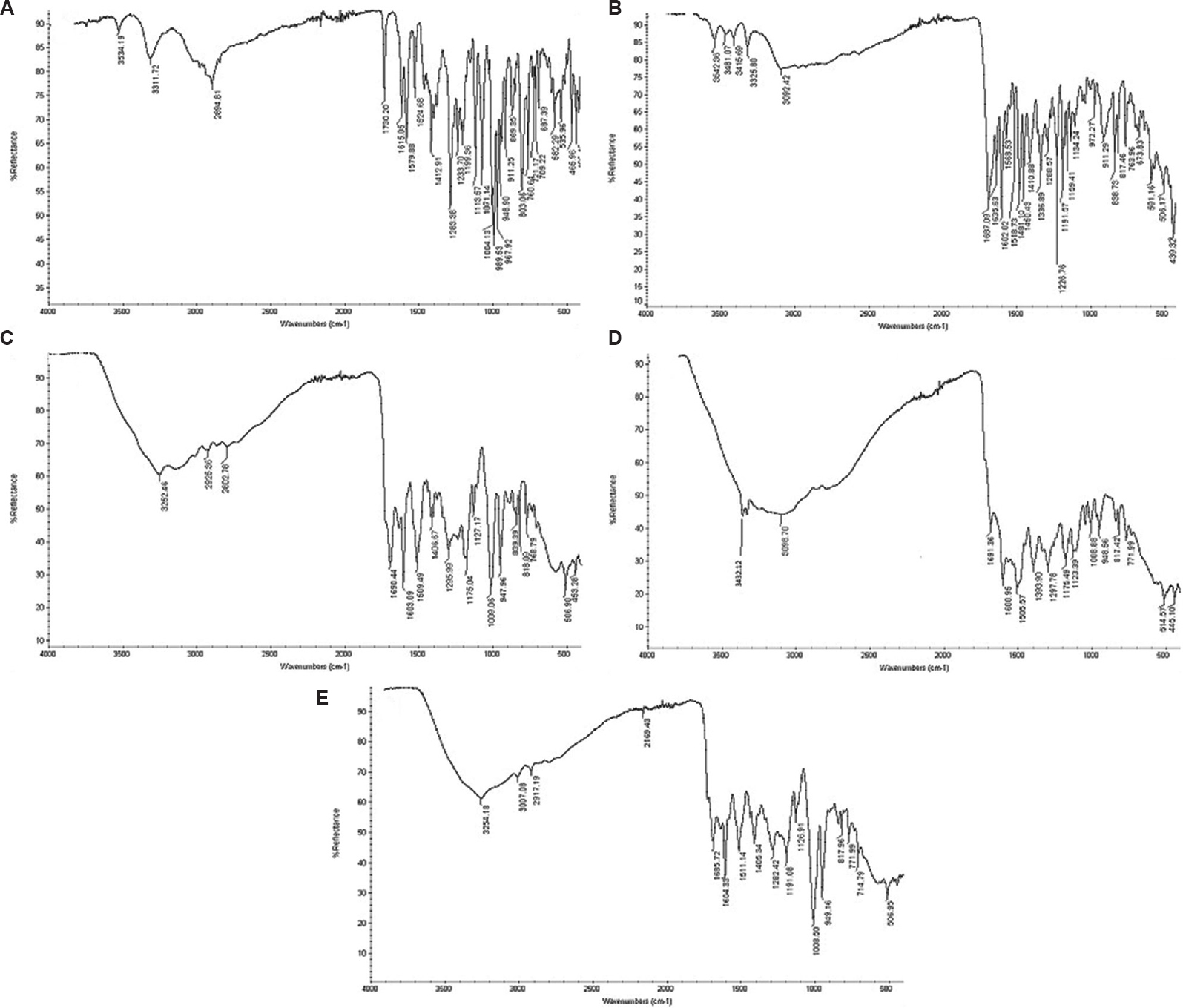
- Fourier transform infrared spectroscopy spectra: (A) DOX, (B) FA, (C) FAA, (D) FAH, (E) FAD. FAD, folic acid-doxorubicin; FAA, folic acid–aminocaproic acid acetate; FAH, folic acid–aminocaproic acid hydrazide; DOX, doxorubicin; FA, folic acid.
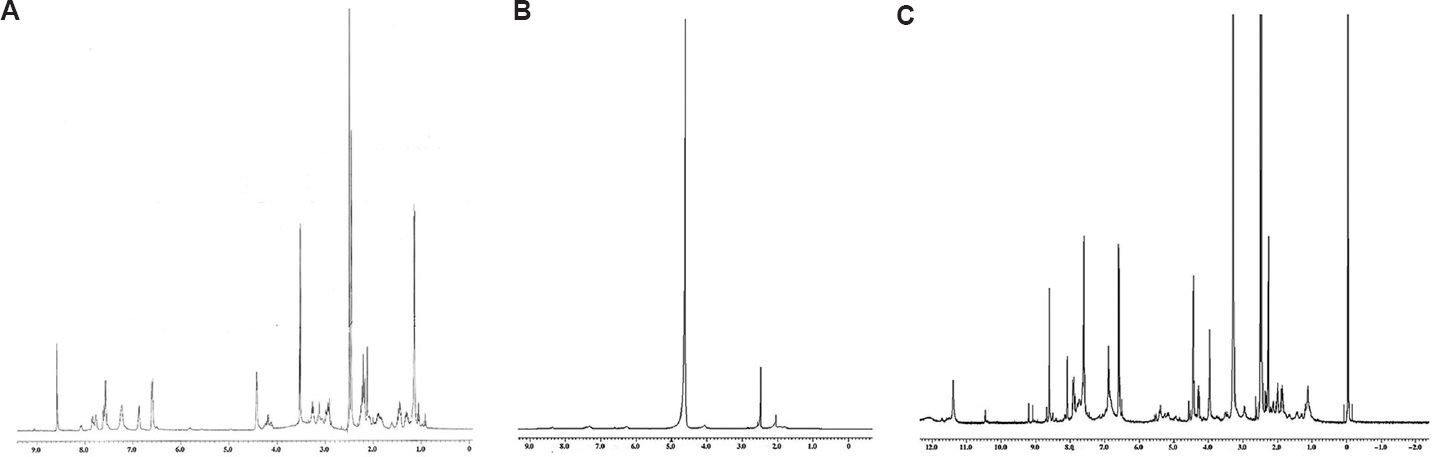
- Proton nuclear magnetic resonance spectroscopy spectra: (A) FAA, (B) FAH, (C) FAD.
Drug release from the conjugate: Drug release from the FAD was determined in three different buffers, i.e. sodium acetate buffer (pH 5.0), phosphate buffer (pH 6.5) and phosphate-buffered saline (PBS, pH 7.4), using the dialysis bag method16. FAD (2 mg) was taken in a dialysis bag (MW, 3500 Da, HiMedia, Mumbai) and dispersed in 20 ml of buffer. The whole assembly was kept over a magnetic stirrer (Remi Equipments, Mumbai), 100 rpm at 37°C. At 0.25, 0.5, 0.75, 1, 2, 3 and 4 h time intervals, samples (0.5 ml) were taken out for drug release analysis and replaced with the same volume of fresh buffer. The released amount of DOX from FAD was determined by ultraviolet (UV) spectrophotometer (Cintra, Japan) at λmax of 480 nm.
Preparation of solid lipid nanoparticles (SLNs): DOX-loaded SLNs (SLN-D) and FAD conjugate-loaded SLNs (SLN-C) were prepared separately by the solvent injection method as reported previously with modifications17. Lipid phase containing DOX/FAD was prepared by melting tristearin:HSPC:DSPE:cholesterol at a concentration 10 mg/ml in a molar ratio (1:1.5:1:1.2) at 70°C in ethanol. The aqueous phase was prepared by dissolving 0.5 per cent tween 80 in 20 ml PBS (pH 7.4). Lipid phase and an aqueous phase both were warmed at 70°C. Lipid phase was gradually added (25 drops min−1) into the aqueous phase using syringe (22-gauge needle speed) under high-speed stirring (2000 rpm) using a magnetic stirrer (Remi Equipments). The resulted lipid suspension was sonicated by a probe sonicator (PCI, India). The SLN-D and SLN-C were concentrated by centrifugation at 20,000 rpm for 30 min and again suspended separately in fresh 20 ml PBS (pH 7.4) separately, containing one per cent of tween 80. The suspended SLNs were again stirred for 1 h on magnetic stirrer at ambient temperature. Unentrapped DOX/FAD was removed by using Sephadex G-50 minicolumn.
Characterization of solid lipid nanoparticles (SLNs): Particle size, polydispersity index (PDI) and zeta potential: The average particle size, polydispersity index (PDI) and zeta potential of the SLN-D and SLN-C formulations were determined in distilled water as a dispersion medium using photon correlation spectroscopy (PCS) with a Zeta sizer (Malvern Instruments, UK), equipped with the Malvern PCS software (Zetasizer Nano software v3.30, Malvern Panalytical, UK).
Entrapment efficiency and per cent drug loading: The amount of entrapped DOX/FAD was estimated by the method reported by Sun et al18, 2019. The SLN-D and SLN-C formulations (free from unentrapped DOX/FAD) were lysed by Triton X-100 (0.1% v/v) and filtered. Absorbance of the filtrate was read at 480 nm and 307 nm for DOX and FAD, respectively, using a UV spectrophotometer (Cintra, Japan). Per cent of DOX/FAD entrapment and per cent drug loading were calculated by using formula shown below.

Morphology of solid lipid nanoparticles (SLNs): The shape of the SLN-D and SLN-C formulations was determined by transmission electron microscopy (TEM; Philips, Tecnai 20, Holland) and scanning electron microscopy (SEM; JEOL- JSM-T330A, Japan). Samples were prepared by diluting the suspension of both the formulations with distilled water and stained with one per cent solution of phosphotungstic acid in water.
In vitro drug release and release kinetic study: In vitro drug release from the SLN-D and SLN-C formulations were determined by a diffusion method using a dialysis bag with slight modifications16 (MW, 3500 Da, HiMedia). SLN-D and SLN-C formulations suspension (containing the drug and conjugate equivalent to 5 mg) free from any unentrapped DOX/FAD was filled in a dialysis bag, sealed and suspended in 50 ml PBS (pH 7.4) taken in a beaker with continuously stirring at a constant speed. The temperature was maintained at 37°C±2°C. At regular time intervals, i.e. 1, 2, 4, 6, 8, 12, 24 and 48 h, samples (2 ml) were withdrawn to determine absorbance using UV spectrophotometer (Cintra, Japan) at λmax of 480 nm and 307 nm for SLN-D and SLN-C, respectively. Simultaneously, the volume was replaced with the same volume of buffer solution. The obtained data were fitted to different kinetic models, i.e. zero-order, first-order, Korsmeyer–Peppas and Higuchi models. The correlation coefficient was determined for each model.
Cytotoxicity and cellular uptake studies: The cytotoxicity of DOX, FAD, SLN-D and SLN-C against brain cancer cells was assessed by the sulforhodamine blue (SRB) assay19. For the cytotoxicity study, U87 MG cells were procured from the National Centre for Cell Sciences (NCCS), Pune, India, and cultured. The cultured cells were incubated with the formulations in final drug concentrations, i.e. 0.1 µg/ml, 1 µg/ml, 10 µg/ml, 100 µg/ml at standard conditions, respectively for 48 h. The viability of cells was expressed in terms of percentage compared to untreated cells (control).
Cellular uptake study was also performed on U87 MG cells using confocal microscopy (Genexplorer diagnostic and Research Pvt. Ltd., Ahmedabad). U87 MG cells were cultured in Petri dishes by taking the same medium as used for the cell cytotoxicity study, until density reached up to 80 per cent confluence. The medium was replaced with plain coumarin 6 (C6) and coumarin 6-loaded SLNs (SLN C6) and incubated for 2 h. After incubation, cells were fixed and washed using PBS (pH 7.4). Then, 10 μl of 5 mg/ml propidium iodide (PPI) was added to stain nucleus. After 30 min, stain cells were again washed and fluorescence was observed under confocal microscope. The same procedure was followed to study the uptake after four hours. The samples and PPI-stained cell nucleus were found to be in blue colour and red colour, respectively.
Statistical analysis: The data obtained from the experiments were analyzed by one way analysis of variance, lack of fit tests and multiple correlation coefficients. Student’s t test was used to test the statistical significance wherever applicable. Obtained data were expressed as mean±standard deviation (n=3).
Results & Discussion
In the present study, SLN-D and SLN-C coated with tween 80 were prepared to enhance delivery of DOX to the brain for the treatment of brain cancer. The conjugate, FAD was synthesized sequentially including synthesis of intermediates such as FAA, FAH and FAD. For the synthesis of the FAA, FA and AMA acetate were used. The FA has α- and γ-carboxylic acid and both groups can be activated in the presence of DCC/NHS which acts as a catalyst. However, because of the high activity of γ-carboxylic acid, it preferentially conjugates with a free amino group of AMA acetate with the formation of the amide bond. This amide bond formation was confirmed by the appearance of a peak at 3252.6 cm−1 (N-H stretching of amide) in the FTIR spectra of the FAA. The spectra also contain a peak at 1739.12 cm−1 indicating the C=O stretching of the ester group of FAA. Some other peaks at 1690.44 cm−1 (C=O stretching of amide) and 1505.57 cm−1 (aromatic C=C) also confirmed the synthesis of the FAA (Table I and Fig. 2).
| Wave number (cm−1) | Functional group | Strength |
|---|---|---|
| FAA | ||
| 3252.6 | N-H stretching (amide bond) | Medium |
| 1739.12 | C=O stretching (ester group) | Strong |
| 1690.44 | C=O stretching (amide bond) | Strong |
| 1505.57 | C=C stretching (aromatic C=C) | Strong |
| FAH | ||
| 3432.12 | N-H stretching (amine) | Medium |
| 3098.70 | C-H stretching | Medium, broad |
| 1691.36 | C=O stretching | Medium |
| 1505.57 | Aromatic C=C stretching | Medium |
| FAD | ||
| 3254.18 | N-H stretching | Weak, broad |
| 3007.08 | C-H stretching (alkanes) | Medium |
| 2917.19 | O-H stretching | Medium, broad |
| 1730.20 | C=O stretching (ketone) | Strong |
| 1685.72 | C=O stretching (amide) | Strong |
| 1652.12 | C=N stretching (imine) | Weak |
| 1190.08 | C-O stretching (ether) | Medium |
FAD, folic acid-doxorubicin; FAA, folic acid–aminocaproic acid acetate; FAH, folic acid–aminocaproic acid hydrazide
In the next step, FAH was synthesized by the hydrazinolysis of the FAA in alcoholic solution. The appearance of peak at 3432.12 cm−1 (N-H stretching), 3098.70 cm−1 (C-H stretching), 1691.36 cm−1 (C=O stretching), 1600.95 cm−1 (N-N stretching) and 1505.57 cm−1 (aromatic C=C) confirmed the synthesis of hydrazide, i.e. FAH.
In the last step, FAD was synthesized by FAH and DOX. During the synthesis, the electrophilic carbon atom of a ketone group present in DOX was targeted by the nucleophilic atom of the FAH amine group. The reaction resulted in the formation of a -C=N- bond. The peak at 1652.12 cm−1 confirmed the formation of –C=N- bond. Peak at 1191.08 cm−1 due to -C-O-C- stretching (ether) also confirmed the conjugation of DOX with FAH. The appearance of other peaks at 3254.18 cm−1 (N-H stretching), 2917.19 cm−1 (O-H stretching), 3007.08 cm−1 (C-H stretching) and 1685.72 cm−1 (C=O stretching of amide) also confirmed the synthesis of FAD. The FAD synthesis was also confirmed by 1H NMR spectroscopy (Bruker ACF300, Germany) (Fig. 3).
The 1H NMR spectrum of FAD was recorded on an instrument operating at a frequency of 500 MHz using DMSO d6 solvent. Signals at 2.9, 2.5 and 2.2 confirmed the presence of AMA moiety, the characteristic signals appeared at 8.6, 7.6, 6.8, 6.6, 4.4 and 4.29 confirmed the conjugation of FA moiety to AMA and the presence of signals at 8.0, 7.5, 4.2 and 1.19 ppm confirmed the conjugation of DOX with AMA.
The 1H NMR spectrum of FAD (Fig. 3) also showed that during conjugation, one mole of FA reacted with one mole of DOX as the height of the signals due to FA and DOX, i.e. at 8.6 ppm and at 7.5 ppm, are nearly the same15.
The release of DOX from the FAD was determined in acidic buffer (pH 5.0 and 6.5) to simulate the acidic environment of cancer cells20,21, while PBS (pH 7.4) was used to simulate physiological conditions. The results of drug release studies showed the highest rate of drug release at pH 5 and it was 48±0.5 per cent in four hours. However, at pH 6.5 and 7.4, it was 9.4±0.3 per cent and 8.2±0.2 per cent, respectively in four hours. The reason for the highest release of DOX at low pH may be due to the presence of >C=N- linkage in FAD which hydrolyzed in an acidic environment. On the basis of release studies, it was confirmed that after internalization of FAD into the cancer cells, the DOX would be released in the acidic lysosomal pH (4.5-5.0). The drug release profile from the conjugates at different pH is shown in Fig. 4.
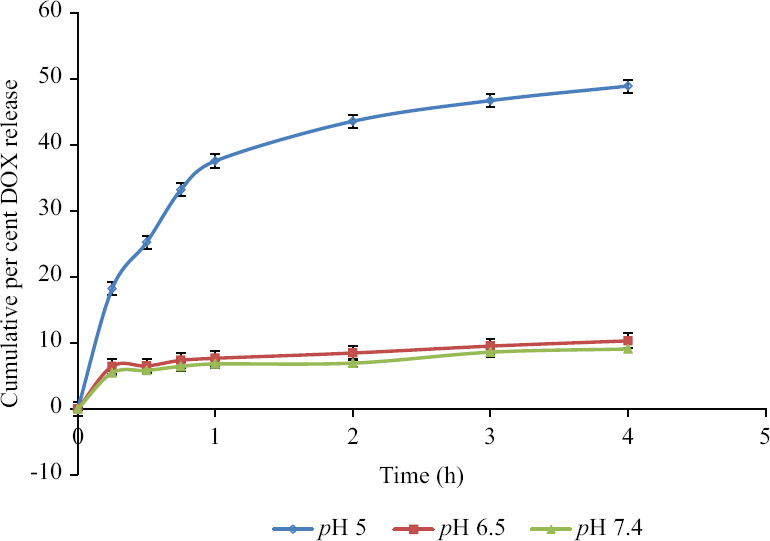
- Cumulative per cent release of DOX at pH 5, 6.5 and 7.4 (n=3). DOX, doxorubicin
SLN-D and SLN-C were prepared by the mixing of lipids, i.e. tristearin, HSPC, DSPE. The triglyceride lipid tristearin and cholesterol constituted the core of the SLNs, while phospholipids HSPC and DSPE formed the periphery of the SLNs. During SLNs formation, ethanol travels across the ethanol–lipid phase in to the aqueous phase. The evaporation of ethanol at 70°C caused rigidization of the lipid phase, leading to the formation of SLNs. The free drug and the entrapped drug in SLNs possess different weights. Due to high molecular weight, SLNs pass through the column unhindered, without penetrating the gel matrix, whereas smaller molecules or free drug get retarded in the column. The SLN-D and SLN-C were characterized for the particle size, PDI, zeta potential, entrapment efficiency and surface morphology (Table II). The average particle size of SLN-D was found to be 205.9±1.2 nm with 45.0±0.2 entrapment efficiency and 40.2±0.5 per cent drug loading of the DOX, respectively. On the other hand, the particle size of the SLN-C was found to be 220.4±2.2 nm with 36.2±0.6 entrapment efficiency and 35.3±0.3 per cent drug loading of the conjugate, respectively. The slight increase in particle size of SLN-C than SLN-D may be due to entrapment of the FAD molecule having higher molecular weight than DOX. This may also be a reason for low entrapment efficiency (%) and per cent drug loading (%) of the SLN-C than SLN-D. The zeta potential (to determine the stability) of SLN-D and SLN-C was found to be −30.2±1.43 and −29.3±1.26 mV, respectively. Since, particles with a slight negative charge repel and reduce the aggregation potential, resulting in improved stability22, the value of zeta potential of both the study formulations, were found to be in a range which indicated sufficient stability.
| Formulations | Particle size (nm) | PDI | Zeta potential (mV) | Entrapment efficiency (%) | Loading efficiency (%) |
|---|---|---|---|---|---|
| SLN-D | 205.9±1.2 | 0.2630±0.05 | −30.2±1.43 | 45.0±0.2 | 40.2±0.5 |
| SLN-C | 220.4±2.2 | 0.2422±0.03 | −25.3±1.26 | 36.2±0.6 | 35.3±0.3 |
SLN, solid lipid nanoparticles; PDI, polydispersity index
To check the homogeneity in the particle size, PDI was determined. The PDI values of SLN-D and SLN-C were found to be less than 0.3, which confirmed the narrow size distribution of SLNs in dispersion. The surface morphology of prepared formulations was observed by TEM and SEM. The images (Figs 5 and 6) clearly revealed that the SLNs possess spherical shape with smooth surface.

- Transmission electron microscopy images of (A) SLN-D and (B) SLN-C. SLN-D, DOX-loaded SLNs, SLN-C, FAD conjugate-loaded SLNs; SLNs, solid lipid nanoparticles.
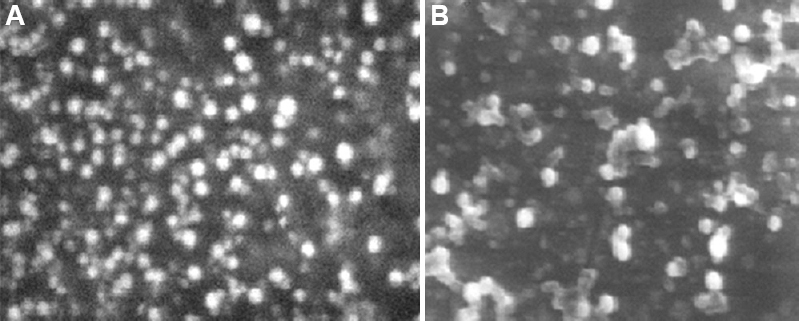
- Scanning electron microscopy images of (A) SLN-D and (B) SLN-C. SLN-D, DOX-loaded SLNs.
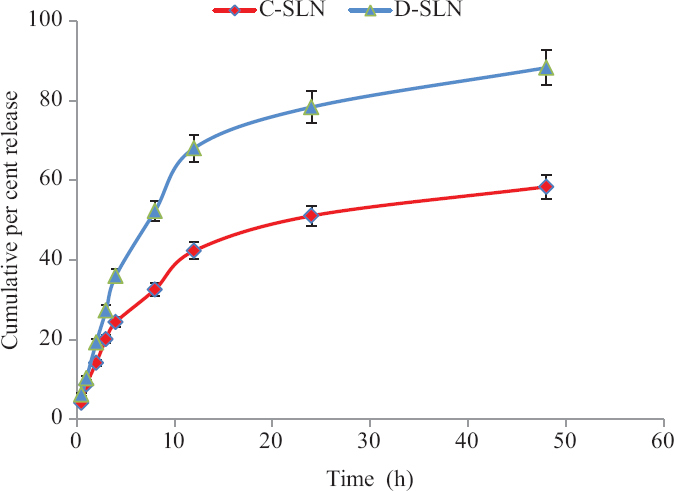
The in vitro release of DOX and FAD from SLNs when determined (pH 7.4) with respect to time (Fig. 7), it was found that 88.3±1.9 per cent of DOX and 58.4±1.6 per cent of FAD were released from SLN-D and SLN-C, respectively, in 48 h. The release kinetics of DOX and FAD from the SLNs was analyzed for zero-order, first-order, Korsmeyer-Peppas and Higuchi model and was found to be best fitted to the Higuchi model as R2 values for DOX and FAD were found to be 0.9890 and 0.9420, respectively (
-
In vitro drug release of the DOX and FAD (n=3). P *≤0.05
| Pharmacokinetic model | DOX | FAD | ||
|---|---|---|---|---|
| Regression coefficient value (R2) | Release rate constant (k) | Regression coefficient value (R2) | Release rate constant (k) | |
| Zero order | 0.8650 | 16.09 | 0.7520 | 13.83 |
| First order | 0.9960 | 1.967 | 0.8390 | 1.935 |
| Korsmeyer-Peppas model | 0.9420 | 0.715 | 0.7150 | 0.756 |
| Higuchi model | 0.9890 | 0.059 | 0.9420 | 2.577 |
DOX, doxorubicin; FAD, folic acid-doxorubicin
The cytotoxicity of the plain DOX, FAD, SLN-D and SLN-C was determined by SRB assay using U87 MG cell line. The plain DOX, FAD, SLN-D and SLN-C showed IC50 (half maximal inhibitory concentration) value 6.3, 4.2, 3.2 and 2.5 µg/ml, respectively indicating that the plain DOX showed least toxicity towards U87 MG cells in comparison to FAD, SLN-D and SLN-C. This could be due to the P-glycoprotein (P-gp) efflux of plain DOX by the U87 MG cell lines23. The cytotoxicity of FAD was found to be greater than DOX. This could be due to the lipophilic behaviour of FAD which facilitates higher uptake by U87 MG cells. Further, the presence of FA in FAD augmented the internalization in U87 MG cells via FR-mediated endocytosis15. The results also showed that both SLN-C and SLN-D were more cytotoxic as compared to plain DOX and FAD. This could be attributed to the presence of tween 80 coating as a part of SLNs structure as tween 80 is known to cause fluidization of the lipoidal cell membrane resulting in enhanced permeability of SLNs across the cell membrane24,25. Furthermore, the tween 80 inhibits Pgp–related efflux by cancer cells and enhance the receptor mediated transcytosis16,24 to augment the carrier-mediated delivery of chemotherapeutics to the brain. These two possible mechanisms could be the reason behind the higher uptake of SLN-C and SLN-D which is in concurrence with available literature. SLN-C was also found to be more cytotoxic than SLN-D. This could be due to the higher uptake of conjugate FAD through folate receptors (FR) which are overexpressed on cancer cells. On internalization in the cancer cells (target site), the DOX was released from the FAD due to the hydrolysis of the hydrazone bond present between FA-AMA and DOX15. In all the formulations, survival fraction of cells was found to be inversely dependent on the concentration of the drug (Fig. 8). The cytotoxicity study confirmed the higher cytotoxicity of the SLN-C than SLN-D, FAD and plain DOX towards brain cancer cells.
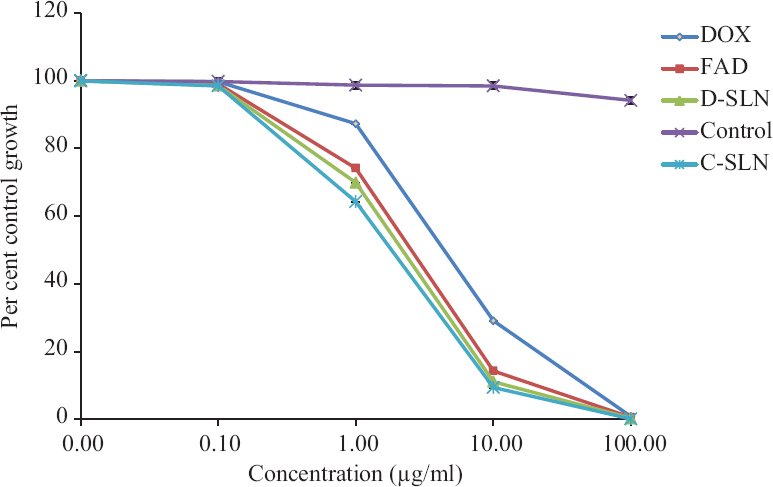
- Cytotoxicity assessed by SRB assay on U87 MG cells after 48 h. The Figure depicts the per cent control growth of U87 MG cells treated with plain DOX, FAD and SLN-C at 0.1 µg/ml, 1 µg/ml, 10 µg/ml and 100 µg/ml doses. Data are represented as the mean±standard deviation (n=3).
To check the internalization of tween 80-coated SLNs in brain cancer cells, the cellular uptake studies were performed on U87 MG cells at different time intervals by using coumarin 6 (C6) as a fluorescent stain. After two hours of incubation, 20±0.5 per cent C6 and 53±0.3 per cent SLNs-C6 were internalized in U87 MG cells which increased up to 25±0.3 and 58±0.2 per cent, for C6 and SLN C6, respectively after tour hours (Fig. 9A and B). The overlay image of SLNs C6 also confirmed the targeting of tween 80-coated SLNs to the U87 MG cells [Fig. 9B(II)] reconfirming the efficiency of the tween 80-coated SLNs in delivering the entrapped drug in U87 MG cells as previously discussed. On the basis of the obtained results, it was concluded that the tween 80-coated SLN-C can effectively deliver the drug to the brain cancer cells. However, the lack of drug release studies using the BBB model was a limitation of this study which should be taken into consideration in the future.

- Confocal micrographs showing the intracellular uptake of SLNs by U87 MG cells. (A) Per cent fluorescence cell after treatment with plain C6 and SLN C6 after 2 h and 4 h. Data are presented as the mean±SD (n=3), (B) U87 MG cells incubated with C6 and SLN C6 for two hours. The nuclei stained by propidium iodide (shows in red), and C6 and SLN C6 is intrinsically blue fluorescent. C6, coumarin 6; SLN C6, coumarin 6-loaded SLNs; SLNs, solid lipid nanoparticles.
Overall, the results suggested the significance of the optimized SLNs in improving the uptake and localization of FAD inside the cancer cells. In short, the prepared SLN-C formulation displayed potential to deliver higher amount of DOX selectively in brain cancer cells, thus providing the targeted therapy for the treatment of brain cancer.
Supplementary Fig. 1
Supplementary Fig. 1 Representation of synthesis FAD. AMA: aminocaproic acid; FA: folic acid; DOX: doxorubicin.Supplementary Fig. 2
Supplementary Fig. 2 In vitro drug release kinetic model of DOX from optimized SLNs. (A) Zero order, (B) first-order, (C) Korsmeyer–Peppas and (D) Higuchi model.Supplementary Fig. 3
Supplementary Fig. 3 In vitro drug release kinetics model of FAD from optimized SLNs. (A) Zero order, (B) first order, (C) Korsmeyer–Peppas and (D) Higuchi model.Acknowledgment:
The author acknowledge the Lipoid, Germany, for providing HSPC and DSPE samples as a gift.
Financial support & sponsorship: This work was supported by the UGC BSR [grant number F-25-1/2013-14(BSR)/7-57/2007(BSR)].
Conflicts of Interest: None.
References
- Doctor-approve patient information from American Society of Clinical Oncology (ASCO). Available from: https://www.cancer.net/cancer-types/brain-tumor/statistics
- Phase 2 study of concurrent radiotherapy and temozolomide followed by temozolomide and lomustine in the treatment of children with high-grade glioma:A report of the Children's Oncology Group ACNS0423 study. Neuro Oncol. 2016;18:1442-50.
- [Google Scholar]
- Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat. 2015;19:1-12.
- [Google Scholar]
- Brain drug delivery by adsorption-mediated transcytosis. In: Gao H, Gao X, eds. Brain targeted drug delivery system. London: Academic Press; 2019. p. :159-83.
- [Google Scholar]
- Transferrin-conjugated magnetic dextran-spermine nanoparticles for targeted drug transport across blood-brain barrier. J Biomed Mater Res A. 2017;105:2851-64.
- [Google Scholar]
- Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. 2018;135:311-36.
- [Google Scholar]
- Effect of surfactant coating on brain targeting polymeric nanoparticles;a review. Indian J Pharm Sci. 2018;80:215-22.
- [Google Scholar]
- Development of nanoparticles for drug delivery to brain tumor:The effect of surface materials on penetration into brain tissue. J Pharm Sci. 2019;108:1736-45.
- [Google Scholar]
- Surface engineered polymeric nanocarriers mediate the delivery of transferrin-methotrexate conjugates for an improved understanding of brain cancer. Acta Biomater. 2015;24:140-51.
- [Google Scholar]
- Enhanced brain targeting of temozolomide in polysorbate-80 coated polybutylcyanoacrylate nanoparticles. Int J Nanomedicine. 2011;6:445-52.
- [Google Scholar]
- Potential of solid lipid nanoparticles in brain cancer treatment. Res Pharm. 2017;1:11-20.
- [Google Scholar]
- Colorectal cancer-targeted delivery of oxaliplatin via folic acid-grafted solid lipid nanoparticles:Preparation, optimization, and in vitro evaluation. Artif Cells Nanomed Biotechnol. 2018;46:1236-47.
- [Google Scholar]
- Improving breast cancer therapy using doxorubicin loaded solid lipid nanoparticles:Synthesis of a novel arginine-glycine-aspartic tripeptide conjugated, pH sensitive lipid and evaluation of the nanomedicine in vitro and in vivo. Biomed Pharmacother. 2019;116:109006.
- [Google Scholar]
- Synthesis of a new pH-sensitive folate-doxorubicin conjugate and its antitumor activity in vitro. J Pharm Sci. 2013;102:530-40.
- [Google Scholar]
- Curcumin loaded nanostructured lipid carriers:In vitro digestion and release studies. Polyhedron. 2019;164:113-22.
- [Google Scholar]
- Transferrin-tailored solid lipid nanoparticles as vectors for site-specific delivery of temozolomide to brain. J Nanopart Res. 2013;15:1518.
- [Google Scholar]
- Mesoporous silica nanoparticles as a delivery system for improving antiangiogenic therapy. Int J Nanomedicine. 2019;14:1489-501.
- [Google Scholar]
- Sulforhodamine B colorimetric assay for cytotoxicity screening. Nature Protocols. 2006;1:1112-16.
- [Google Scholar]
- Spectrin conjugated PLGA nanoparticles for potential membrane phospholipid interactions:Development, optimization and in vitro studies. J Drug Deliv Sci Technol. 2020;60:102087.
- [Google Scholar]
- Manipulating extracellular tumour pH:An effective target for cancer therapy. RSC Adv. 2018;8:22182-92.
- [Google Scholar]
- pH-sensitive polymeric nanoparticles for tumor-targeting doxorubicin delivery:Concept and recent advances. Nanomedicine (Lond). 2014;9:487-99.
- [Google Scholar]
- A mechanism for overcoming P-glycoprotein-mediated drug resistance:Novel combination therapy that releases stored doxorubicin from lysosomes via lysosomal permeabilization using Dp44mT or DpC. Cell Death Dis. 2016;7:e2510.
- [Google Scholar]
- Modeling of ultra-small lipid nanoparticle surface charge for targeting glioblastoma. Eur J Pharm Sci. 2018;117:255-69.
- [Google Scholar]
- Polysorbate 80 coated solid lipid nanoparticles for the delivery of temozolomide into the brain. Open Pharmacol J. 2018;8:21-8.
- [Google Scholar]




