
Translate this page into:
Rapid molecular identification of a rare β-globin gene deletion & its clinical implication
*For correspondence: anitahnadkarni@yahoo.com
-
Received: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Sir,
β-thalassaemia is one of the most common inherited monogenic disorders prevalent in India, with an overall frequency of 3-4 per cent and an estimate of 10,000-12,000 children born every year as β-thalassaemia major1. This emphasizes on the precise identification of β-thalassaemia carriers and characterization of the β-globin gene mutation as the same will be required for prenatal diagnosis. The standard test for detecting a typical β-thalassaemia carrier is the elevated HbA2 level which ranges from 4.0 to 6.5 per cent2. However, some atypical cases show an elevated HbA2 level >7.0 per cent, wherein the large deletion is suspected. Identification of large deletions in the β-globin gene cluster has recently gained importance because of its significance in unsolved thalassaemia cases and disease prevention. However, conventional diagnostic tests [covalent reverse dot blot (CRDB) hybridization, amplification-refractory mutation system (ARMS) polymerase chain reaction (PCR) and β-globin gene sequencing], targeting point mutations and small insertions–deletions in the β-globin gene fail to detect large deletions3. Thus, a differential approach combining techniques such as southern blotting, quantitative fluorescent (QF) PCR, fluorescent in situ hybridization, multiplex ligation-dependent probe amplification (MLPA) and gap-PCR could be utilized4. In the present study, a combined strategy of MLPA and simplified gap-PCR technique was used to detect a rare deletion of 4056 bp which was mainly detected in couples from the Gujarat region of India.
The study was approved by the Institutional Ethics Committee, ICMR-National Institute of Immunohaematology (NIIH), and the samples were obtained after obtaining written informed consent from the patients. Five families with a history of β-thalassaemia were referred for haemoglobinopathy workup and eventually for prenatal diagnosis during January 2016 to March 2020. Preliminary screening by analyzing the complete blood count and HbA2 and HbF levels by high performance liquid chromatography (HPLC) was carried out. Genomic DNA was extracted and β-globin gene analysis was carried out. MLPA was performed using SALSA MLPA Kit P102 HBB (MRC-Holland, Amsterdam, The Netherlands)5, which contained 29 probes for the 73 kb region of the β-globin gene cluster targeting the locus control region, coding genes in the cluster, the intergenic sequences and 10 reference probes5. The manufacturer’s protocol was followed. The MLPA products were separated on an automated DNA sequencing machine (Applied Biosystems, Genetic Analyzer 3130xl, ThermoFisher Scientific, USA), and the results were analyzed using GeneMapperTM software version 4.0 (ThermoFisher Scientific, USA) and Coffalyser. Net software (Coffalyser.Net - MRC, Holland). Further, a simplified single tube gap-PCR was performed with the help of previously reported primers sequences6. The gap-PCR programme employed to detect the deletion was as follows: initial denaturation at 94°C for five minutes, followed by 30 cycles of denaturation: 94°C for one minute, annealing at 58°C for one minute, extension at 72°C for one minute and a final extension at 72°C for five minutes.
In five families, both or either of the parents (total 7) had an elevated A2 in the range of 7.5-8.8 per cent (mean±SD; HbA2: 7.95±0.45%). In these patients, initial molecular screening by CRDB hybridization, ARMS and β-globin gene sequencing was carried out at ICMR-NIIH, Mumbai and was found to be normal. Out of the five families, four were referred to our centre for prenatal diagnosis as well. In families 1 and 3, one parent had typical beta-thalassaemia indices and DNA analysis identified IVS1-5 (G→C) mutation (HBB: c.92+5G>C) [NM_000518.5 (HBB): c.92+5G>C]. The other parent had high A2, 7.5-8.8 per cent. Each of the above families had a child presenting with severe anaemia requiring frequent blood transfusions. Reverse dot blot analysis of both the affected children showed the presence of HBB: c.92+5G>C homozygous pattern, however, the prenatal report was given (family 1 and 3) as the chorionic villus sample showed the absence of HBB: c.92+5G>C mutation. In the second family, both the parents had high HbA2 and they had a clinically presenting child, who required regular blood transfusion (they had come for prenatal diagnosis). CRDB analysis and whole β-globin gene sequencing showed a normal pattern. As the β-globin gene mutation was undetermined, the patient was called for cordocentesis (family 2). On HPLC analysis of this cordocentesis sample, the HbA2 level of foetal blood was 2.6 per cent, by ruling out maternal contamination this was suggestive of β-thalassaemia minor and the report was issued. Similarly, in family 4, both the parents had raised HbA2 levels, suggesting that they were β-thalassaemia carriers and they were referred for prenatal diagnosis. Family 5 was referred for molecular diagnosis of β-globin gene mutation, as the mother had a raised HbA2 level. In the individuals, where the HbA2 was found to be elevated, deletion in the β-globin gene was suspected, that was being missed by the conventional diagnostic methods. Hence, with the standardization of MLPA technique, these uncharacterized samples were analyzed again. In MLPA, probe amplification spanning from the δβ intergenic region, β-globin promoter, exon 1, 2, up to the intron 2 of the β-globin gene was found to be reduced in the individuals with elevated HbA2 and their affected children (Fig. 1). The results of MLPA analysis helped in designing a gap-PCR which generated two bands in heterozygous condition: an internal control band of 449 bp and a mutant band of 1127 bp; and a single band each of 449 bp and 1127 bp under normal and homozygous condition for the deletion, respectively (Fig. 2A). Further, DNA sequencing of the mutant band showed a deletion of 4056 bp that extends from 2.7 kb downstream of the δ-globin gene to IVS-2 of the β-globin gene (Fig. 2B). Table shows the haematological and molecular details of the families studied. We observed that the individuals heterozygous for 4056 bp deletion had microcytic anaemia and the β-thalassaemia homozygous patients for 4056 bp deletion presented with a severe clinical condition requiring regular blood transfusion.
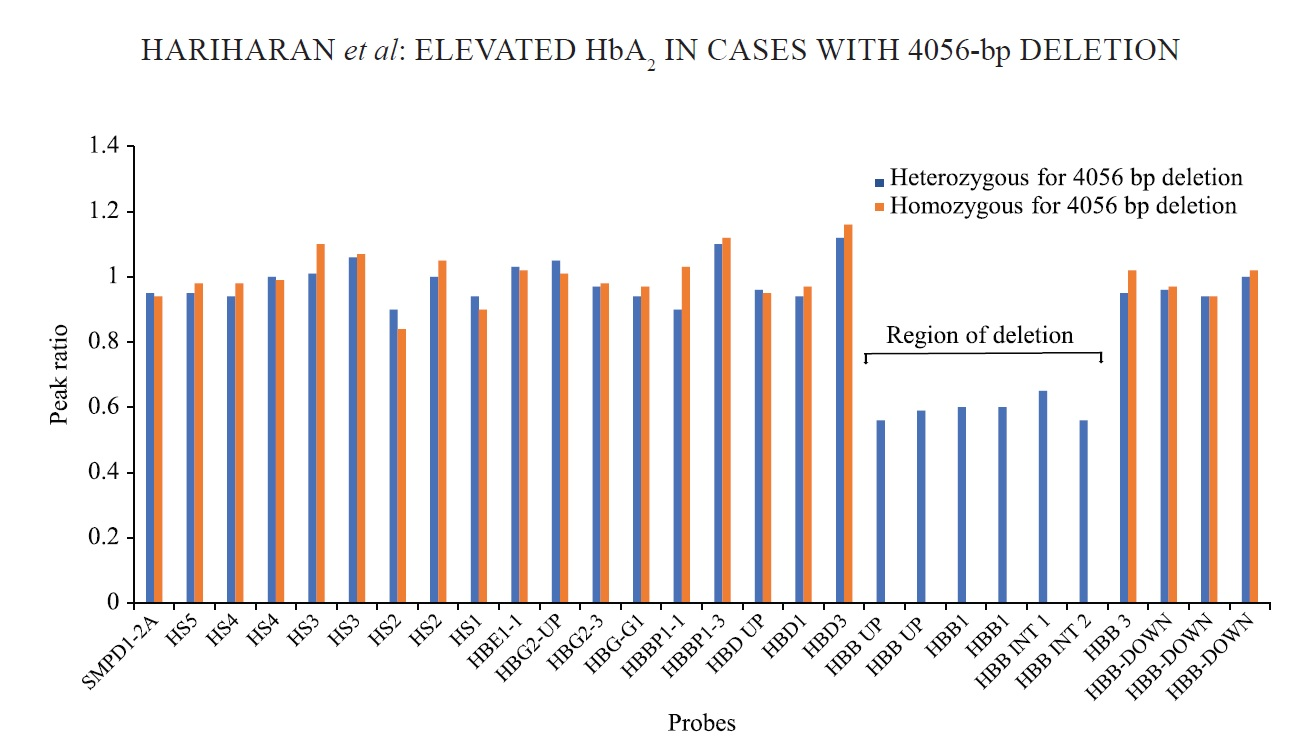
- MLPA analysis of two different patient samples. β-globin cluster showing genomic regions showing 4056 bp deletion in heterozygous (blue) and homozygous (red) patients. The Y axis represents the probe dosage ratio and the X axis represents the MLPA probes. MLPA, Multiplex ligation-dependent probe amplification.
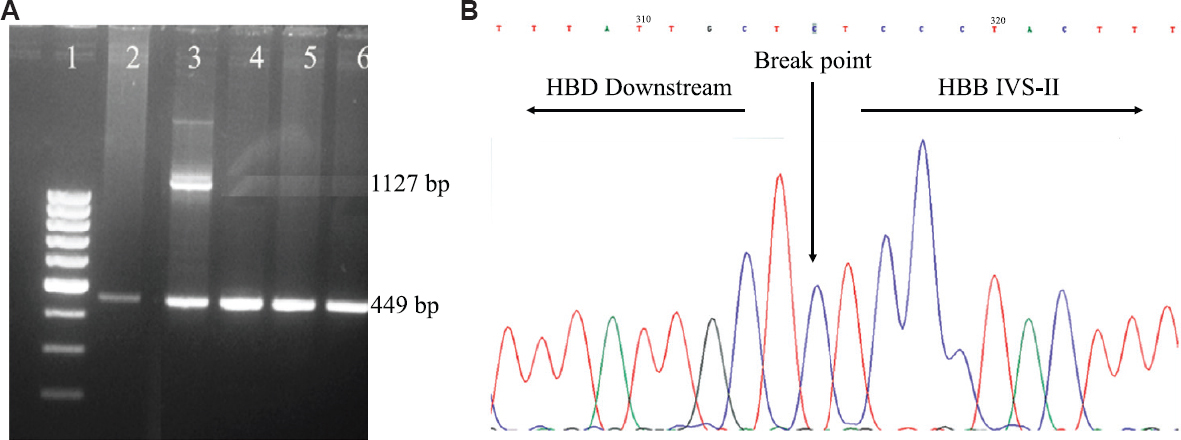
- (A) 1.5 per cent agarose gel showing amplified products of gap-PCR. Lane 1: 100 bp ladder. Lane 2, 4, 5, 6: Normal control, showing the presence of control band of 449 bp. Lane 3: Sample heterozygous for 4056 bp deletion showing an internal control band of 449 bp and a mutant band of 1127 bp. (B) Automated DNA sequencing of the patient harbouring 4056 bp deletion in heterozygous condition. The electropherogram shows the breakpoint position of the 4056 bp deletion.
| Age | Caste | Origin | RBCs (106/µl) | Hb (g/dl) | MCV (fl) | MCH (pg) | HbA2 (%) | HbF (%) | β-globin genotype by conventional technique (CRDB) | β-globin genotype after MLPA and gap-PCR | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family 1 | |||||||||||
| Father | 23 yr | Rajput | Saurashtra | 5.23 | 10.5 | 61.4 | 20.1 | 5.0 | 0.1 | HBB: c. 92+5G>C Heterozygous | HBB: c. 92+5G>C Heterozygous |
| Mother | 22 yr | Rajput | Saurashtra | 4.50 | 9.8 | 63.6 | 21.8 | 8.1 | 3.8 | Normal | 4056 bp deletion Heterozygous |
| Affected child* | 3 yr | Rajput | Saurashtra | 4.70 | 12.0 | 74.6 | 20.6 | 2.1 | 3.2 | HBB: c. 92+5G>C Homozygous pattern | HBB: c. 92+5G>C +4056 bp deletion |
| Foetus (CVS) | 12 wk gestation | Rajput | Saurashtra | - | - | - | - | - | - | HBB: c. 92+5G>C Absent | Follow up |
| Family 2 | |||||||||||
| Father | 30 yr | Rajput | Rajkot | 6.19 | 12.2 | 61.6 | 19.7 | 7.5 | 1.1 | Normal | 4056 bp deletion Heterozygous |
| Mother | 28 yr | Rajput | Rajkot | 4.13 | 8.3 | 62.2 | 20.1 | 8.8 | 2.8 | Normal | 4056 bp deletion Heterozygous |
| Affected child* | 4 yr | Rajput | Rajkot | 4.89 | 12.9 | 75.5 | 26.4 | 3.2* | 4.6 | Not amplified | 4056 bp deletion homozygous |
| Foetus (AF) | 17 wk gestation | Rajput | Rajkot | - | - | - | - | - | - | Normal | 4056 bp deletion heterozygous |
| Family 3 | |||||||||||
| Father | 28 yr | Patel | Surat | 5.58 | 11.8 | 69.4 | 21.1 | 8.2 | 3.2 | Normal | 4056 bp deletion Heterozygous |
| Mother | 25 yr | Patel | Surat | 5.10 | 10.2 | 64.5 | 20.0 | 5.0 | 0.7 | HBB: c. 92+5G>C Heterozygous | HBB: c. 92+5G>C Heterozygous |
| Affected child* | 5 yr | Patel | Surat | 3.81 | 11.3 | 88.7 | 29.7 | 3.2 | 4.0 | HBB: c. 92+5G>C Homozygous pattern | HBB: c. 92+5G>C +4056 bp deletion |
| Foetus (CVS) | 12 wk gestation | Patel | Surat | - | - | - | - | - | - | HBB: c. 92+5G>C Absent | Follow up |
| Family 4 | |||||||||||
| Father | 32 yr | Ahir | No blood samples provided, only DNA samples were referred along with HbA2 and HbF levels | 7.5 | 2.0 | Normal | 4056 bp deletion Heterozygous | ||||
| Mother | 28 yr | Ahir | 8.05 | 3.2 | Normal | 4056 bp deletion Heterozygous | |||||
| Foetus (AF) | 17 wk gestation | Ahir | - | - | Normal | 4056 bp deletion Heterozygous | |||||
| Family 5 | |||||||||||
| Father | 34 yr | Singh | No blood samples provided, only DNA samples were referred along with HbA2 and HbF levels | 2.9 | 0.6 | Normal | Normal | ||||
| Mother | 30 yr | Singh | 7.5 | 2.1 | Normal | 4056 bp deletion Heterozygous | |||||
*Post-transfused sample. CVS, chorionic villus sample; AF, amniotic fluid; RBCs, red blood cells; Hb, haemoglobin; HbA2, haemoglobin A2; HbF, foetal Hb; MCV, mean corpuscular volume; MCH, mean corpuscular haemoglobin; MLPA, multiplex ligation-dependent probe amplification; CRDB, covalent reverse dot blot; HBB, Hemoglobin subunit beta
β-thalassaemia occurs due to mutation in the β-globin gene which leads to reduced or absent β-globin chain synthesis. In rare cases, the deletional form of β-thalassaemia, involving variable-sized deletion of the β-globin gene cluster region with an exceptionally high HbA2 level with mild elevation in HbF level, is observed7. In this study, a 4056 bp deletion was identified in heterozygous form in seven individuals showing high HbA2 levels (7.5-8.8%) with slight elevation in the HbF levels (1.1-3.8%). Furthermore, two affected cases of compound heterozygous for 4056 bp deletion and HBB: c.92+5G>C β-globin gene mutation, and one affected case homozygous for 4056 bp deletion were identified in this study. The molecular characterization of 4056 bp deletion showed that the deletion extended from the 3157 bp 5’ upstream from the cap site to the 3’-IVS-2 region of the β-globin gene. Previous studies have reported around 15 deletions in different populations that remove a variable genomic segment from the β-globin gene and result in β-thalassaemia phenotype8. Popovich et al9, 1986, characterized a β-globin gene deletion of 4237 base pairs, which showed a similar extent of deletion from 3.3 kb upstream from the cap site to the middle of IVS-2 of the β-globin gene in a family of Czechoslovakian descent with higher HbA2 levels ranging from 8.1 to 9.0 per cent and HbF levels ranging from 3.3 to 5.5 per cent. Dimovski et al10, 1993, detected a 1605 bp deletion of the 5’ beta-globin promoter region leading to β0-thalassaemia in a Croatian family with unusually high HbA2 (7.6-8.2%) with HbF levels: 5.8-8.5 per cent. Similarly, Gallienne et al11, 2010, identified a novel deletional mutation of 909 base pairs in the β-globin gene, which involved the removal of the β-globin gene promoter, exon 1, IVS-I entirely and most of exon 2 of the β-globin gene (−478 to +432) with exceptionally high HbA2 phenotype (HbA2: 8.6-9.0%; HbF: 1.7-3.9%). In the Indian population, the most common β-globin deletional mutation observed is the 619 bp deletion; however, these patients also show an average HbA2 level of 6.5 per cent, slightly higher than the typical β-thalassaemia12. Thein13, 2013, reported that the deletional mutations removing the 5’ end of β-globin gene and promoter sequences involving TATA, CCAAT and CACCC boxes, which are involved in regulating the transcription, may result in unusually higher HbA2 level and mild elevation in the HbF level. A similar observation is found in the present study as well. The plausible mechanism of raised HbA2 and HbF levels may be due to the increased interaction of the locus control region with the cis-δ-globin and γ-globin promoter region in the absence of competing β-globin gene, thus enhancing their expression13. Waye et al14, 2017, also characterized two novel deletions of 538 bp and 1517 bp which removed the β-globin gene promoter, 5’ UTR and exons 1 and 2. Majority of the deletional mutation of β-thalassaemia span from the 5’ promoter region of the gene to the β-IVS-2 region at the 3′ end which could be attributed to the presence of a recombinational hot spot.
Large deletions in the β-globin cluster escape traditional diagnostic techniques6. In our study as well, the CRDB, ARMS PCR and direct DNA sequencing failed to identify the presence of 4056 bp deletion. During routine diagnosis, as the HbA2 level in the presence of the deletion is comparatively higher than the average HbA2 value for β-thalassaemia trait, a deletion in the β-globin gene can be easily suspected. A combined strategy of MLPA, QF-PCR and gap-PCR was thus used to detect this deletion. Previously, the 4056 bp deletion was first identified by Mayuranathan et al6, 2012, in a patient of Gujarat origin. They used combined MLPA and PCR techniques to identify this deletion wherein the PCR generated only one deletion-specific band of 1.1 kbp. Hence, heterozygosity or homozygosity for the deletion could not be distinguished. In this study, we have described a fast, reliable, relatively cost-effective gap-PCR approach to identify 4056 bp deletion that generates an amplification product of 449 bp in the presence of a normal chromosome, and a 1.1 kb deletion specific band, which can easily distinguish between a normal, heterozygous and homozygous condition of the deletion.
The molecular characterization of large deletional mutations in the β-globin gene cluster is extremely critical for at-risk couples seeking prevention as these deletions escape identification and compromise the prenatal diagnosis in laboratories that follow the conventional methodology to detect β-globin gene mutations. The 4056 bp deletion seems to be a region-specific mutation, hence gap-PCR could be easily performed, instead of going for an expensive technique; however, if the finding of gap-PCR are negative then MLPA, QF-PCR can be performed in suspected cases.
Acknowledgment
Authors acknowledge all the clinicians who did the foetal sampling procedures over the years and all the previous staff and students for their technical support.
Financial support & sponsorship: This study was funded by the Indian Council of Medical Research, New Delhi, India.
Conflicts of Interest: None.
References
- Burden of thalassemia in India:The road map for control. Pediatr Hematol Oncol. 2017;2:79-84.
- [Google Scholar]
- Hemoglobin A2 (HbA2) has a measure of unreliability in diagnosing β-thalassemia trait (β-TT) Curr Med Res Opin. 2018;34:945-51.
- [Google Scholar]
- The changing trends in prenatal diagnosis of hemoglobinopathies in India:The quest of a single center to reduce the burden of disease over three decades. Hemoglobin. 2021;45:112-18.
- [Google Scholar]
- Multiplex ligation-dependent probe amplification screening of isolated increased HbF levels revealed three cases of novel rearrangements/deletions in the beta-globin gene cluster. Br J Haematol. 2010;148:154-60.
- [Google Scholar]
- Nine unknown rearrangements in 16p13.3 and 11p15.4 causing alpha- and beta-thalassaemia characterised by high resolution multiplex ligation-dependent probe amplification. J Med Genet. 2005;42:922-31.
- [Google Scholar]
- A novel deletion of β-globin promoter causing high HbA2 in an Indian population. Haematologica. 2012;97:1445-7.
- [Google Scholar]
- Two new beta-thalassemia deletions compromising prenatal diagnosis in an Italian and a Turkish couple seeking prevention. Haematologica. 2009;94:1289-92.
- [Google Scholar]
- Filipino beta zero thalassaemia:A high Hb A2 beta zero thalassaemia resulting from a large deletion of the 5'beta globin gene region. J Med Genet. 1993;30:240-4.
- [Google Scholar]
- Molecular characterization of an atypical beta-thalassemia caused by a large deletion in the 5'beta-globin gene region. Am J Hum Genet. 1986;39:797-810.
- [Google Scholar]
- A beta zero-thalassaemia due to a 1605 bp deletion of the 5'beta-globin gene region. Br J Haematol. 1993;85:143-7.
- [Google Scholar]
- Characterization of a novel deletion causing beta-thalassemia major in an Afghan family. Hemoglobin. 2010;34:110-4.
- [Google Scholar]
- The spectrum of beta-thalassaemia mutations on the Indian subcontinent:The basis for prenatal diagnosis. Br J Haematol. 1991;78:242-7.
- [Google Scholar]
- The molecular basis of β-thalassemia. Cold Spring Harb Perspect Med. 2013;3:a011700.
- [Google Scholar]
- Characterization of two novel deletions involving the 5'region of the β-globin gene. Hemoglobin. 2017;41:239-42.
- [Google Scholar]




