
Translate this page into:
Molecular characterization of varicella zoster virus isolated from clinical samples in India
For correspondence: Dr Pragya D. Yadav, Maximum Containment Facility, Maximum Containment Laboratory, ICMR-National Institute of Virology, Sus Road, Pashan, Pune 411 021, Maharashtra, India e-mail: hellopragya22@gmail.com
-
Received: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
Varicella zoster virus (VZV) strains are classified into six different clades based on the sequencing of its genome. Clades 4 and 5 are reported from India based on the single-nucleotide polymorphism (SNP). Till now, multiple clade circulations using partial sequences have been reported from India due to the lack of availability of the full VZV genome sequence. This study conducted a genome sequencing of VZV in India to identify circulating clade.
Methods:
Four clinical samples obtained from symptomatic patients tested positive for VZV by real-time PCR were used. These four samples were preferred to retrieve the genomic VZV sequence using the next-generation sequencing method. A reference-based assembly method was used to retrieve the genome of VZV, which was further analyzed.
Results:
At the least, 98 per cent of the whole-genome sequences were recovered from the four samples. The VZV sequences obtained in this study formed a separate monophyletic branch with clade 5, indicating it to be evolved from a distinct ancestor. The nucleotide-based analysis revealed 13 different SNP mutations and one multiple nucleotide variation in the VZV sequences when compared to one of the clade 5 genomes having accession number: DQ457052.1.
Interpretation & conclusions:
The present study described approximately 98 per cent of the genome sequence of VZV from India. The availability of these genomic sequences will lead to enrichment in the clinical genomic data set from India. The available data would help in the development of diagnostic methods along with evolutionary analysis. We hypothesize the existence of a new sub-clade that belongs to clade 5 and propose further experiments to confirm these results.
Keywords
Clade
genome
next-generation sequencing
phylogenetics
single-nucleotide polymorphism
varicella zoster virus

Varicella zoster virus (VZV) is an alpha-herpesvirus and a causative agent of chickenpox (varicella) and shingles (zoster). Chickenpox is the primary infection characterized by fever and a generalized rash, which is prevalent in children. Although a live-attenuated vaccine has been available since 19741, chickenpox remains a primary childhood infection. Propagation of VZV in cell culture was first reported in 1953, and a complete nucleotide sequence for VZV was first determined using the M13-dideoxynucleotide technology from the Dumas strain2.
The genome of VZV is approximately 125 kb in length and consists of a double-stranded DNA and has 72 known open reading frames (ORFs)2. Phylogenetic analysis of VZV sequences shows that this genetically stable virus shows sufficient variation to classify it into six clades. Restriction fragment length polymorphism (RFLP) in ORF38, ORF54 and ORF62345 has been the most common method employed for virus classification. Another method classifies the virus based on scattered single-nucleotide polymorphisms (SNPs) in ORF1, ORF21, ORF50 and ORF54, thereby classifying the VZV in four main viral clades, namely A, B, C and J6. Classification of VZV by glycoprotein sequences has led to another system of classification into four significant clades designated as A, B, C and D4. The fourth system of VZV classification refers to ORF22-, ORF21- and ORF50-based genotyping and classifies the virus into five confirmed clades E1, E2, J, M1 and M2, and two provisional clades M3 and M47. Classification of VZV based on complete genome leads to six significant clades that have a distinct geographic distribution pattern89. Based on the full genome sequence-based classification, clade 2 is found to be dominantly present in Asia4. However, in countries belonging to the South Asian subcontinent, namely India, Pakistan, Bangladesh and Sri Lanka, clades 4 and 5 are found to be more prevalent56.
The genetic diversity among VZV sequences is 0.00063, which is almost 40 times less than that of other large double-stranded DNA viruses such as herpes simplex virus and cytomegalovirus8. However, with such a small genetic diversity, the clinical condition manifested by VZV is varied1011. A study from India based on clinical-epidemiological details of VZV-infected cases reported symptoms varying from mild fever with a rash to very severe (bleeding manifestation) that led to mortality10. VZV infection causes a communicable disease that mostly spreads through the vesicular skin cell, a place where it is highly concentrated12. Although VZV infections are prevalent across India1314, complete genome sequences from India are not available. The present study was thus aimed to identify the circulating clade of VZV in India using its genomic sequence from clinical samples.
Material & Methods
This study was conducted at Maximum Containment Laboratory of the Indian Council of Medical Research - National Institute of Virology (ICMR-NIV), Pune, India during April 2017. The samples used in this study were received as part of the focal VZV outbreaks that had occurred in Jaipur, Rajasthan, and Mumbai, Maharashtra, India. Written informed consent was taken before the collection of the clinical specimen from VZV-infected patients. The study was approved by the Institute’s Human Ethics Committee.
In the earlier published work, 79 samples from Maharashtra (n=50), Gujarat (n=12) and Rajasthan (n=17) States, India, were tested by real-time PCR to test the VZV positivity15. Sixty one positive clinical samples were observed from these three locations15. However, based on the availability of the sample and volume required to generate complete genomic sequences, using next-generation sequencing (NGS), two samples each from Rajasthan and Maharashtra were selected for this study.
Next-generation sequencing (NGS):
DNA library preparation: The whole-genome sequencing of VZV was performed using the NGS platform. DNA was extracted from 1 ml of the VZV-positive samples using a Qiagen DNA extraction kit (Qiagen, Hilden, Germany). The extracted DNA was quantified using Qubit 2.0 (Invitrogen, Carlsbad, USA) and diluted to 0.2 ng/μl. The diluted DNA was used to prepare the DNA library by Nextera DNA library preparation (Illumina, California, USA) using the previously described protocol16. The DNA library was quantified using the KAPA library quantification kit (Kapa Biosystems, Roche, Basel, Switzerland). The final DNA libraries to be sequenced were normalized and loaded onto the MiniSeq mid-output cartridges (300 cycles) for paired-end sequencing using the Illumina MiniSeq platform, (Illumina, USA).
The library for DNA sample with accession number MH499466 was made using NEBNext® Ultra™ DNA Library Prep Kit for Illumina® (New England Biolabs® Inc. Massachusetts, USA) to retrieve more sequences. The library was made as per the manual of the kit with slight modifications. The final input concentration of the DNA sample for NGS library preparation was 100 ng. The concentration was decided based on the previous optimization performed using different DNA concentrations. Six PCR cycles were used for the amplification of adaptor-ligated DNA.
Analysis of the reads: The FastQ files generated from the Illumina machine were imported to CLC Genomics Workbench v10.1.1 (Qiagen, Aarhus, Denmark) for analysis. Initially, contiguous sequences (contigs) were generated using the de novo contig assembly method, as implemented in the CLC Genomics Workbench. These contigs were identified using the BLAST programme to be similar to the clade 5 sequences. VZV sequence belonging to clade 5 (accession number DQ457052.1) was downloaded as a reference sequence from GenBank. Further, reference-based mapping was performed to retrieve the complete genome of VZV from the FastQ files. These sequences were submitted to the public repository.
Phylogenetic analysis: Complete reference sequences for VZV genomes were downloaded from the GenBank database to generate the phylogenetic tree (Table I). These sequences were aligned and manually checked for its correctness. The difference in the nucleotide and the per cent identity was retrieved using CLC Genomics Workbench. Maximum likelihood tree was generated using GTR+ G+I as the nucleotide substitution model in MEGA version 7.0 software17. A bootstrap replication of 1000 cycles was used to assess the significance of the phylogenetic tree generated.
| Accession number | Clade |
|---|---|
| DQ479954.1 | Clade 1 |
| DQ479953.1 | Clade 1 |
| DQ479958.1 | Clade 1 |
| DQ479961.1 | Clade 1 |
| DQ479959.1 | Clade 1 |
| AY548171.1 | Clade 1 |
| NC_001348.1 | Clade 1 |
| AY548170.1 | Clade 1 |
| DQ674250.1 | Clade 1 |
| EU154348.1 | Clade 1 |
| AB097932.1 | Clade 2 |
| AB097933.1 | Clade 2 |
| DQ008354.1 | Clade 2 |
| DQ008355.1 | Clade 2 |
| DQ479955.1 | Clade 3 |
| AJ871403.1 | Clade 3 |
| DQ479956.1 | Clade 3 |
| DQ479957.1 | Clade 3 |
| DQ479960.1 | Clade 4 |
| DQ452050.1 | Clade 4 |
| JN704705.1 | Clade 5 |
| JN704707.1 | Clade 5 |
| DQ457052.1 | Clade 5 |
| JN704706.1 | Clade 5 |
| JN704708.1 | Clade 5 |
| JN704704.1 | Clade 5 |
| JN704709.1 | Clade 8 |
| JN704710.1 | Clade 9 |
Results
Next-generation sequencing: Reference-based mapping using the reference genome [DQ457052.1 (CA123)] led to the retrieval of 124,700 to 121,928 bp of VZV sequences from different clinical samples (Table II). The Q30 was >95 per cent for the reads generated from the clinical samples. VZV sequences having accession numbers MH499466 and MH499467 were collected from Jaipur, Rajasthan, and MH499469 and MH499468 were collected season from Mumbai, Maharashtra. Samples with accession numbers MH499468 and MH499467 had partial genome (~97.7-98.5% genome coverage) retrieval due to the availability of lesser sample volume. The details regarding the sample type, total read generated and length of the sequences recovered along with the percentage of the reads mapped are given in Table II.
| Accession number | Sample type | Total reads | Viral reads (% read mapped) | Length of VZV sequence (bp) (124,771-125,239) | Per cent coverage |
|---|---|---|---|---|---|
| MH499469 | Vesicle fluid | 3,451,352 | 52,772 (1.5) | 123,646 | 99.09 |
| MH499468 | Vesicle fluid | 3,458,492 | 4747 (0.13) | 121,928 | 97.73 |
| MH499467 | Lung tissue, skin, heart blood | 3,815,536 | 10,480 (0.27) | 122,945 | 98.54 |
| MH499466 | Vesicular fluid | 2,425,108 | 1,787,961 (73.27) | 124,700 | 99.94 |
VZV, varicella zoster virus
Phylogenetic analysis: Fig. 1A and B depicts the phylogenetic tree for the VZV sequences that have genomic coverage of more than 99 and 97 per cent, respectively, to reference sequence. The analysis revealed that the Indian sequences belonged to clade 5. However, an interestingly separate branch/cluster was observed with bootstrap support of above 90 and 80 per cent for the two different trees generated (Fig. 1A and B). We also determined the differences and the per cent identity of the VZV sequences analyzed in the study (Fig. 2). The upper right matrix and a lower left matrix of Fig. 2 denote the nucleotide differences and per cent identity, respectively, between the VZV reference and Indian VZV sequences obtained from Indian samples. It was observed that VZV sequences belonging to clade 5 had nucleotide per cent identity that distinctly formed two different groups, except MH499468, which might be due to its smaller length. Further, the nucleotide difference of strain CA123 (accession number: DQ457052.1) belonging to clade 5 and Indian sequences with other reference VZV sequences was also significant.
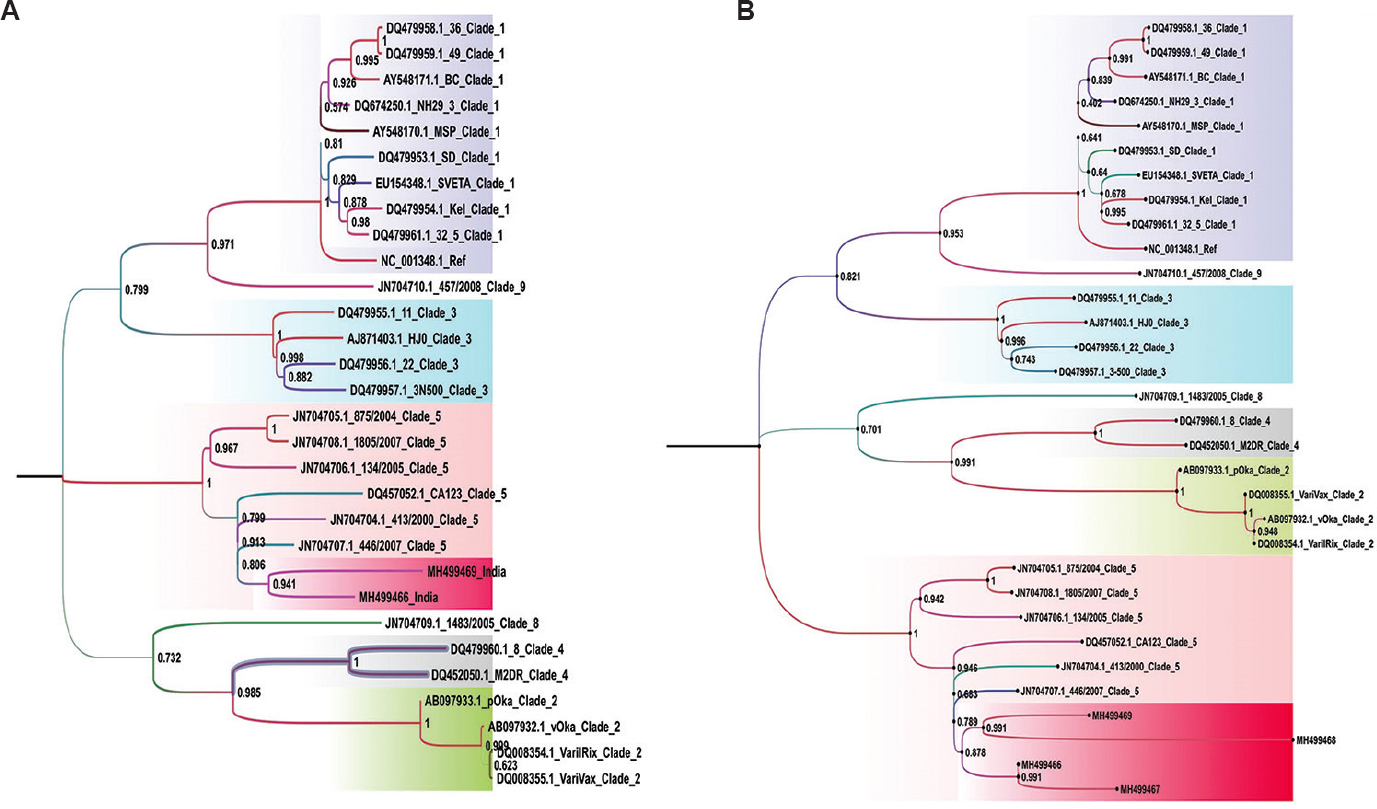
- A maximum likelihood evolutionary tree for varicella zoster virus genomic coverage of more than 99 per cent (A) and more than 97 per cent (B) was drawn using the general time-reversible model, having a gamma parameter as 0.1000 with an invariable rate variation of 49.64 and 49.63, respectively. A bootstrap replication of the 1000 cycles was performed to assess the statistical robustness.
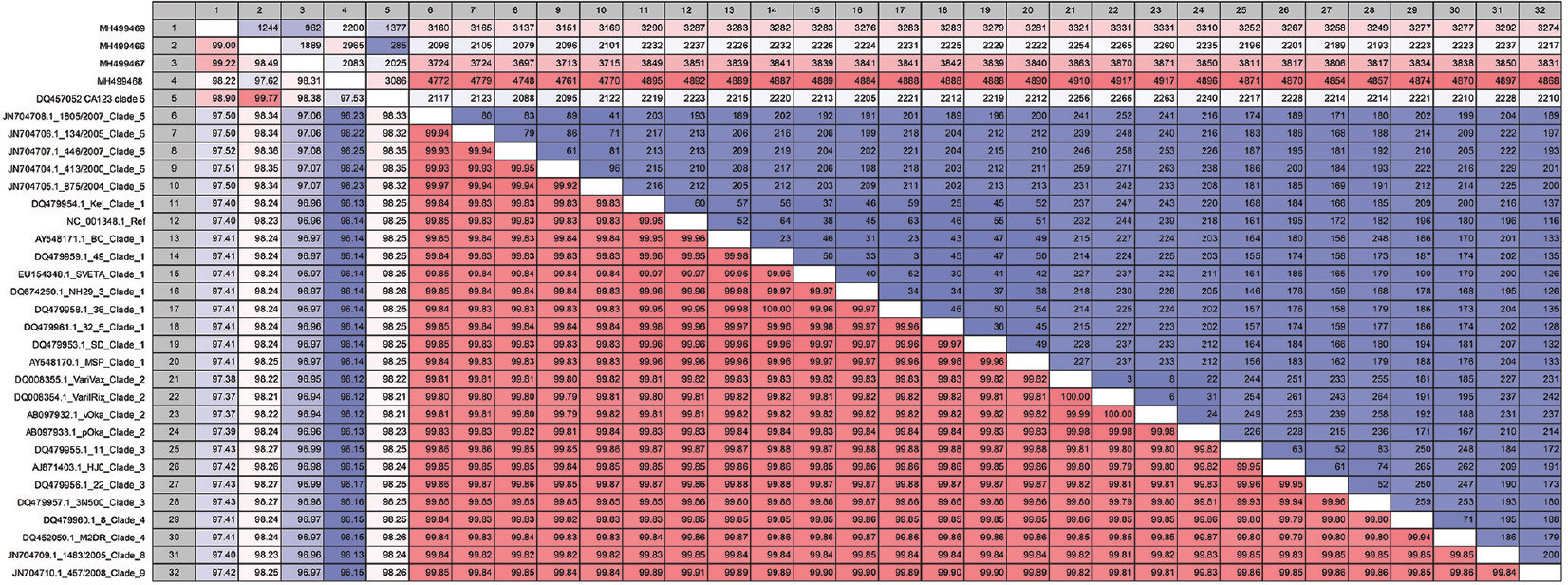
- The nucleotide differences and per cent identity between the varicella zoster virus sequences used in this study obtained from the CLC Genomics Workbench. Nucleotide difference is shown in the upper right panel and per cent identity on the left lower panel.
Table III enlists the 13 SNPs observed in the Indian VZV five sub-clade compared to reference clade 5 genomes (accession no.: DQ457052.1). Three of the 13 SNPs (A72484C, G74974A and C75130T) and a single multi-nucleotide polymorphism (MNP) (CC75126-75127TT) were observed in the ORF40 region that encodes for major capsid protein. Of the remaining nine SNPs, non-synonymous amino acid changes were observed for four SNPs (Table III).
| Position of nucleotide | Region | Amino acid | Type | Reference sequence (DQ457052) | Allele | |
|---|---|---|---|---|---|---|
| Indian sequence | Reference sequence | |||||
| 5473 | ORF6/primase | L | L | SNV | A | G |
| 36,730 | ORF22/tegument protein | V | I | SNV | A | G |
| 46,940 | ORF27/nuclear matrix protein | D | D | SNV | T | C |
| 55,694 | ORF30/terminase subunit | P | P | SNV | G | A |
| 72,484 | ORF40/major capsid protein | I | I | SNV | C | A |
| 74,974 | ORF40/major capsid protein | L | L | SNV | A | G |
| 75,126-75,127 | ORF40/major capsid protein | AD | VD | MNV | TT | CC |
| 75,130 | ORF40/major capsid protein | D | D | SNV | T | C |
| 76,161 | ORF41/minor capsid protein | I | I | SNV | A | C |
| 77,463 | ORF45/putative ATPase subunit of terminase | Y | Y | SNV | G | A |
| 93,872 | ORF54/portal protein | S | P | SNV | G | A |
| 99,559 | ORF58/unknown | V | V | SNV | T | G |
| 101,040 | ORF58/unknown | M | I | SNV | T | C |
| 113,055 | ORF66/serine-threonine protein kinase | G | C | SNV | T | G |
SNV, single-nucleotide variation; MNV, multiple nucleotide variation; ORF, open reading frame
Discussion
The sequencing of large genomes like VZV is a challenging task. In this study the NGS platform was used to sequence the genome as (i) VZV being a slow-growing virus in tissue culture, the isolations took long time, and (ii) clinical samples were limited. Despite these, nearly complete genomes of VZV were obtained using NGS, albeit with some gaps in the sequences. All the four VZV sequences formed a cluster with clade 5 sequences. The Indian VZV sequences formed a distinct cluster with a separate branch having 70 per cent bootstrap support. This indicated that a different sub-clade belonging to clade 5 was circulating in the Rajasthan and Maharashtra States of India. As the sample size was restricted, the existence of a clade other than the observed sub-clade could not be denied.
Depledge et al18 in their study tried to understand if there was a correlation between viral genetic diversity and disease progression in terms of central nervous system (CNS) and non-CNS cases. They observed that cases with encephalitis showed more considerable genetic variability as compared to VZV-infected cases with meningitis, usually a self-limiting infection. In another study by Peters et al19, it was hypothesized that the virus must have co-evolved along with its host, and the recombination among various strains with the host genome might be responsible for variations specific to a given geographic region. All four genome sequences obtained from India belonged to clade 5, and formed a separate branch, thereby suggesting that there might have been geographical region-specific host interactions leading to various nucleotide changes.
Sequence data from India are fragmented. The present analysis helped to understand the phylogenetic analysis of nearly 98 per cent complete VZV sequences from India and its comparison with other global VZV sequences. The changes in the structure or function of the major capsid protein caused from the change in the SNPs needs to be further experimentally studied. Attempt to correlate disease symptoms and specific sequence variations in the VZV genome was none, apart the earlier published work18. The SNPs observed in this study were from the limited dataset, which was also the limitation of this study, and indicated a need for VZV sequencing from a substantial number of confirmed clinical cases. This will help in understanding the viral diversity associated with the clinical manifestation in the disease condition.
Financial support & sponsorship: This work was supported by the Indian Council of Medical Research - National Institute of Virology, Pune.
Conflicts of Interest: None.
References
- The varicella vaccine. Vaccine development. Infect Dis Clin North Am. 1996;10:469-88.
- [Google Scholar]
- The complete DNA sequence of varicella-zoster virus. J Gen Virol. 1986;67:1759-816.
- [Google Scholar]
- Restriction fragment length polymorphism of polymerase chain reaction products from vaccine and wild-type varicella-zoster virus isolates. J Virol. 1992;66:1016-20.
- [Google Scholar]
- Global identification of three major genotypes of varicella-zoster virus:Longitudinal clustering and strategies for genotyping. J Virol. 2004;78:8349-58.
- [Google Scholar]
- Evolution and world-wide distribution of varicella-zoster virus clades. Infect Genet Evol. 2011;11:1-10.
- [Google Scholar]
- The out of Africa model of varicella-zoster virus evolution:Single nucleotide polymorphisms and private alleles distinguish Asian clades from European/North American clades. Vaccine. 2003;21:1072-81.
- [Google Scholar]
- Complete-genome phylogenetic approach to varicella-zoster virus evolution:Genetic divergence and evidence for recombination. J Virol. 2006;80:9569-76.
- [Google Scholar]
- Phylogenetic analysis of varicella-zoster virus:Evidence of intercontinental spread of genotypes and recombination. J Virol. 2002;76:1971-9.
- [Google Scholar]
- The molecular epidemiology of varicella-zoster virus:Evidence for geographic segregation. J Infect Dis. 2002;186:888-94.
- [Google Scholar]
- VZV:Pathogenesis and the disease consequences of primary infection. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, eds. Human Herpesviruses:Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press; 2007.
- [Google Scholar]
- Varicella zoster virus infection:Clinical features, molecular pathogenesis of disease, and latency. Neurol Clin. 2008;26:675-97. viii
- [Google Scholar]
- Outbreak of chickenpox in a Union Territory of North India. Indian J Med Microbiol. 2015;33:524.
- [Google Scholar]
- Investigation of an outbreak of varicella in Chandigarh, North India, using a real-time polymerase chain reaction approach. Indian J Med Microbiol. 2017;35:417.
- [Google Scholar]
- Clinico-epidemiological investigation on Varicella Zoster Virus indicates multiple clade circulation in Maharashtra state, India. Heliyon. 2018;4:e00757.
- [Google Scholar]
- Identification and phylogenetic analysis of herpes simplex virus-1 from clinical isolates in India. Access Microbiol. 2019;1:e000047.
- [Google Scholar]
- MEGA7:Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870-4.
- [Google Scholar]
- High viral diversity and mixed infections in cerebral spinal fluid from cases of varicella zoster virus encephalitis. J Infect Dis. 2018;218:1592-601.
- [Google Scholar]
- A full-genome phylogenetic analysis of varicella-zoster virus reveals a novel origin of replication-based genotyping scheme and evidence of recombination between major circulating clades. J Virol. 2006;80:9850-60.
- [Google Scholar]




