
Translate this page into:
Cloning, expression & evaluation of potential immunogenic recombinant capsid premembrane protein of West Nile virus
For correspondence: Dr Jyoti S. Kumar, Division of Virology, Defence Research & Development Establishment, Jhansi Road, Gwalior 474 002, Madhya Pradesh, India e-mail: jyotishukla2@yahoo.co.in
-
Received: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
West Nile virus (WNV) is a neurotropic flavivirus that has emerged globally as a significant cause of viral encephalitis. The early confirmatory diagnosis of WNV infections is important for timely clinical management and in areas where multiple flaviviruses are endemic. Diagnosis of WNV infection is primarily based on serodiagnosis, followed by virus isolation and identification. The aim of this study was to develop and evaluate a highly sensitive and specific immunoglobulin M (IgM) ELISA using the recombinant CprM protein (rWNV-CprM) for rapid, early and accurate diagnosis of WNV.
Methods:
The gene coding for the CprM protein of WNV was cloned and expressed in pET 28a vector followed by purification. An indirect IgM microplate ELISA using purified rWNV-CprM protein was optimized having no cross-reactivity with healthy human serum and serum samples obtained from patients with dengue and Japanese encephalitis viruses infection.
Results:
The comparative evaluation of this rWNV-CprM protein-specific IgM ELISA with plaque reduction neutralization test using 105 blood samples collected from patients suspected to have acute WNV infection revealed 98 per cent concordance with sensitivity and specificity of 100 and 97 per cent, respectively.
Interpretation & conclusions:
The recombinant CprM protein-based WNV-specific ELISA reported in this study may be useful for rapid screening of large numbers of blood samples in endemic areas during outbreaks.
Keywords
CprM gene
blood samples
ELISA
plaque reduction neutralization test
recombinant protein
West Nile virus

West Nile virus (WNV) emerged from relative obscurity in 1999 when the first incursion of the virus into North America caused an outbreak of meningoencephalitis leading to seven deaths in the New York City area12. The laboratory diagnosis is based essentially on immunoglobulin M (IgM) ELISA owing to low transient viraemia making virus isolation difficult34. WNV is an envelope, single-stranded, positive-sense RNA virus with a genome of approximately 11 kb. The genome encodes a single polyprotein to yield the following structural proteins: the capsid (C) protein, the pre-membrane protein, and the envelope glycoprotein. These proteins are followed by the non-structural (NS) proteins, NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS556. The primary role of the C protein in the flavivirus lifecycle is in the assembly of the nucleocapsid and its incorporation into nascent virions78910. Serologic confirmation of WNV infection in humans is possible only by virus isolation and the plaque reduction neutralization test (PRNT) assay. However, both PRNT and virus isolation assays require up to a week for completion, and isolation requires viable virus in samples1112. In addition to reverse transcription polymerase chain reaction (RT-PCR), there are more rapid, sensitive and RT-PCR-based assays such as the TaqMan RT-PCR and loop-mediated isothermal amplification methods which are under evaluation with clinical samples1314.
Most of the WNV-specific IgM ELISA test systems are based on the capture principle and rely on the use of whole virus antigens15. Cross-reactivity among flaviviruses may be a major shortcoming with regard to the IgM ELISA used currently for routine investigation. Therefore, it is essential to develop a simple, safe and cost-effective recombinant protein-based IgM ELISA for the detection of WNV infection at early stages with high degree of sensitivity and specificity. Here we report the development and evaluation of a highly sensitive and specific recombinant protein-based IgM ELISA by targeting the CprM gene of WNV. The gene was expressed in Escherichia coli and purified in a single step and yielded 25 mg of pure protein per liter of shake flask culture. The assay was evaluated with a panel of 170 suspected clinical samples. Cross-reactivity was also monitored with serum samples from patients with dengue and Japanese encephalitis (JE) along with serum samples from healthy individuals. The sensitivity, specificity and applicability of this rWNV-CprM-based IgM ELISA for clinical diagnosis of WNV infection are also described in the present study.
Material & Methods
The study was conducted in the division of Virology, Defense Research and Development Establishment, Gwalior, India. The study protocol was approved by the institutional ethics committee.
The WNV (Eg101 strain) used in this study was obtained from the Institute of Tropical Medicine, Nagasaki, Japan. The virus was propagated by regular passaging in Aedes albopictus clone C6/36 cells and titrated by plaque assay in Vero cells in accordance with the standard protocol. Luria Bertani (LB) broth for bacterial media preparation was obtained from HiMedia Laboratories (Mumbai, India); Ni-NTA superflow resin, viral RNA Mini Kit and anti-His antibody were from Qiagen (Hilden, Germany). RT-PCR kit, isopropyl thiogalactoside (IPTG), diaminobenzidine (DAB), tetramethylbenzidine (TMB), 30 per cent hydrogen peroxide (H2O2), kanamycin and all secondary antibody-enzyme conjugates (anti-mouse, anti-rabbit and anti-human horseradish peroxidase) were from Sigma (USA). WN detect™ IgM capture ELISA from PanBio, Brisbane, Australia. Restriction enzymes were from MBI Fermentas (Hanover, MD, USA).
Cloning and expression of CprM gene in expression vector: Viral RNA was extracted from 140 μl of infected culture supernatant according to the manufacturer's instruction (Qiagen, USA). The genomic region coding for CprM protein was amplified using a set of primers [Forward WN-F5'-CACCA TGGAAATGTCTAAGAAACCAGGAGGGCCC-3´ (NcoI) and the reverse primer WN 5´-CACTCGAGCC TCCGGCTACGTCTTGAGTGGCGTGT-3´ (Xho I)]. In-house designed primers were used in the present study; these primers were utilized for confirmation of clones through Sanger nucleotide cycle sequencing. Sequence results were confirmed through BLAST search (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The purified PCR product (CprM gene) was cloned into the pCR4-TOPO cloning vector followed by sub-cloning in pET28a expression vector. Clones were confirmed by colony lysis followed by nucleotide sequencing.
Expression profile and localization of recombinant protein:The expression was optimized using different concentration of IPTG (0.5, 1, 1.5 and 2 mM) and checked at hourly interval for five hours. Following induction, cells were lysed in lysis buffer and analyzed by 10 per cent sodium dodecyl sulphate polyacrylamide electrophoresis (SDS-PAGE)16.
Purification of the recombinant protein under denaturing conditions:
Harvesting inclusion bodies (IBs) pellet: Recombinant clone in BL21 (DE3) cells was inoculated in one litre of LB broth containing 50 μg/ml kanamycin and subjected to growth at 37°C for three hours with constant shaking. When the OD 600 of the culture attained, 0.6 and 1 ml of un-induced culture were submitted for subsequent SDS-PAGE analysis followed by induction with 1 mM IPTG. The induced culture was allowed to grow at 37°C for three hours. The cell pellet was suspended in 80 ml of cell lysis buffer followed by sonication. The supernatant was discarded and the inclusion body (IB) pellet was applied for further use.
Purification and solubilizaion of inclusion bodies (IBs): The IB pellet was suspended in 100 ml of IBs wash buffer (pH 6.0) containing 50 mM PO4, 200 mM NaCl, 5 mM EDTA, 1 M urea and 1 per cent Triton-X100 followed by centrifugation. The supernatant was discarded, and the purified IB pellet was used for solubilization. IB solubilization buffer (pH 8.0) containing 10 mM Tris-Cl, 100 mM NaCl, 100 mM NaH2PO4, 1 mM EDTA and 8 M urea was added to the purified IB pellet and stirred overnight at room temperature (RT). The solubilized IB was centrifuged and the clear supernatant was used for the purification of rWNV-CprM.
Purification of recombinant CprM protein: The recombinant protein was purified by immobilized metal affinity chromatography using a Ni-NTA superflow chelating agarose column17. Initially, the column was equilibrated with 20 ml of IB solubilization buffer (pH 8.0) and later washing of the column with 50 ml of wash buffer, pH 6.0. The protein was eluted by passing 15 ml elution buffer (pH 4.0)18. Each elute was collected in 1 ml fractions and analyzed on 12 per cent SDS-PAGE followed by dialysis. The final protein concentration was 2 mg/ml19.
Western blot analysis: The purified rWNV-CprM protein was run on a 10 per cent SDS-PAGE and was transferred electrophoretically to nitrocellulose membrane for two hours at 35 V20 followed by overnight blocking. These membrane strips were incubated for one hour at RT with different antibodies, viz. anti-His monoclonal antibodies, rabbit hyperimmune serum against WNV and pre-immunized rabbit serum. The strips were incubated separately with goat anti-mouse and anti-rabbit horseradish peroxidase (HRP) conjugate at a dilution of 1:2000 for one hour at RT followed by washing. Finally, strips were developed with DAB and 30 per cent H2O2 in phosphate buffered saline (PBS).
Indirect immunoglobulin M (IgM) microplate ELISA: An indirect microplate IgM ELISA was standardized using the purified recombinant protein. The purified rWNV-CprM protein was diluted to 300 ng/μl in 0.1 M carbonate buffer, pH 9.6, and used for coating in 96-well microtitre plate (100 μl/well) and incubated at 37°C for two hours. The coated wells were washed once with 1× PBS and then blocked with three per cent bovine serum albumin (BSA) in 1× PBS overnight at 4°C. Following washing as above, 100 μl of WN suspected patient serum samples at a dilution of 1:100 made in 1× PBS was added in the wells and incubated for one hour at 37°C. The plate was incubated with anti-human IgM HRP conjugate (1:3000 dilutions in 3% BSA) followed by washing steps. Reaction was developed with 100 μl of TMB substrate and kept at RT for five minutes. The peroxidase reaction was terminated with 100 μl of 1N H2SO4 and the absorbance was read at 490 nm in an ELISA reader.
Evaluation of indirect IgM microplate ELISA: A total of 105 blood samples from patients suspected to have WNV infection collected from Aravind Eye Hospital, Madurai, India, during 2010-2011 were screened in this study. Serum samples separated from blood were used for various experiments. The cross-reactivity study was also attempted with a panel of serum samples obtained from 20 healthy persons in addition to 20 and 10 samples from dengue/chikungunya patients. The correlation, sensitivity, specificity, positive predictive value and negative predictive values were calculated. The comparative evaluation of rWNV-CprM protein-specific IgM ELISA with PRNT was determined.
Plaque reduction neutralization test (PRNT):Neutralizing antibodies in the samples were measured by 90 per cent PRNT90 according to standard protocol21 with some modifications, using Vero cells. Briefly, heat-inactivated serum samples serially twofold diluted starting from 1:5 to 1:640 and rabbit anti-WN serum in 1:100 and 1:200 dilutions were mixed with 1000 plaque forming units of virus and incubated at 37°C for 60 min. This virus-serum mixture was inoculated (200 μl/well) to Vero cell monolayer cultured on 24-well tissue culture plate along with suitable virus and cell controls and was allowed to adsorb in a five per cent CO2 incubator for two hours at 37°C. Following adsorption, 1 ml of 1.25 per cent methylcellulose overlay medium was added to each well, and the cells were incubated at 37°C in five per cent CO2 incubator for five days. After washing with PBS, cells were fixed with methanol and stained with 0.02 per cent crystal violet solution. The neutralizing antibody titre was expressed as a reciprocal of the dilution of serum, which caused a 90 per cent reduction of plaque formation compared to the plaque number in the virus control.
Results
Expression and purification of recombinant protein: A 620 bp DNA fragment encoding rWNV-CprM protein was amplified by RT-PCR using specifically designed primers. This amplified product was cloned into pET28a vector and transformed in E. coli DH5α (Fig. 1A). The recombinant clone was transformed in E. coli BL 21. Clones were confirmed by PCR and cycle sequencing (Fig. 1B). Induced protein band of 23 kDa was observed at 1 mM IPTG for three hours (Fig. 2A). Purification of the rWNV-CprM protein was performed under denaturing condition. The protein was eluted with the pH gradient and was found to be 95 per cent pure, by gel analysis. The yield of pure rWNV-CprM protein was estimated to be 25 mg/l of shake flask culture. Western blot analysis accomplished with two different antibodies revealed specific 23 kDa protein band, representing the rWNV-CprM. The pre-immunized control serum did not generate any signal (Fig. 2B).
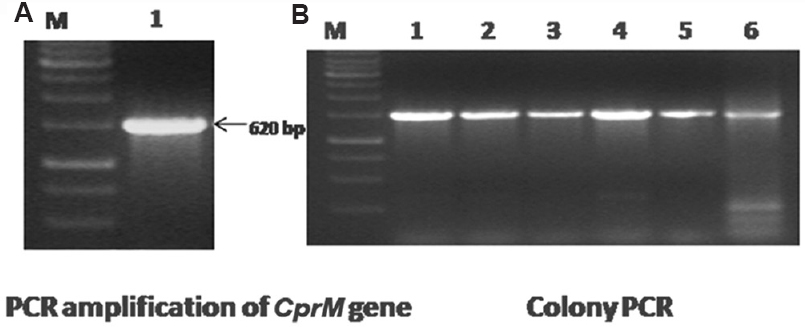
- (A) Polymerase chain reaction amplification of the WNV- CprM gene. Lane M, 1 kb DNA ladder; lane 1, amplified product of 620 bp of the CprM gene. (B) Confirmation of recombinant clones by colony lysis polymerase chain reaction. Lane M, 1 kb DNA ladder; lanes 1-6, amplified products of 620 bp represent recombinant CprM clone.
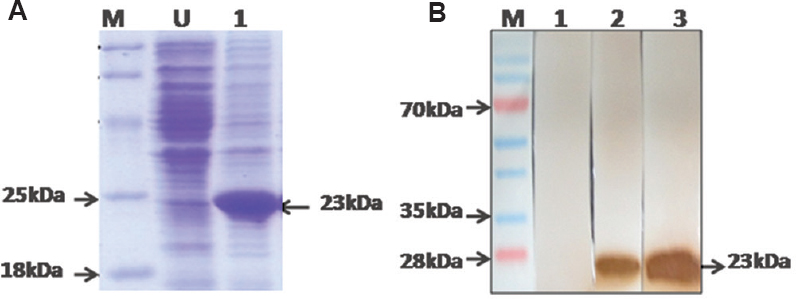
- (A) Sodium dodecyl sulphate polyacrylamide gel electrophoresis analysis of rWNV-CprM protein expression. Lane M, unstained protein marker; lane U, uninduced recombinant clone; lane 1, induced recombinant clone. (B) Western blot analysis of the recombinant protein showing reaction at approximately 23 kDa. Lane M, prestained protein marker; lane 1, reaction with pre-immunized control rabbit serum; lane 2, showing their action with the anti-West Nile rabbit antibody; lane 3, showing the reaction with the anti-His antibody.
Evaluation of recombinant protein-based indirect IgM ELISA:Evaluation of 170 clinically suspected blood samples collected from Aravind Eye Hospital during 3-7 days of illness from patients (35-37 yr) having symptoms of fever, fatigue, joint pain, uveitis and neuroretinitisshowed that 35 samples for chikungunya and 30 were positive for dengue IgM antibody. Subsequently, all the 170 blood samples including those that were dengue and chikungunya positive were processed for WNV detection by IgM capture ELISA. Of the 105 samples, 35 samples were positive for IgM antibodies by indirect ELISA employing recombinant CprM antigen. There was no cross-reactivity observed with dengue and JE virus along with serum from healthy persons. All the positive samples revealed a higher OD value of 0.8-2.00 as compared to 0.1 in the case of negative samples as revealed through colour intensity. None of the serum from healthy individual as well as culture supernatant showed any visual colour reaction. In most of the WN-positive samples, the OD values were in the range of 1.0-2.0 during the acute phase of illness.
Comparative evaluation with plaque reduction neutralization test (PRNT):In vitro neutralization of WNV by the samples was assayed by a PRNT90. Thirty three of 105 samples were found to neutralize 90 per cent of the virus up to 1:160 dilution. The parameter of comparative evaluation with PRNT indicated accordance of 98 per cent with a sensitivity and specificity of 100 and 97 per cent, respectively. In terms of positive and negative predictive values, rWNV-CprM ELISA was found to be in the range of 94-100 per cent as compared to PRNT, respectively. No cross-reactivity was observed between serum samples of healthy persons and those of patients with dengue, JE and chikungunya.
Discussion
WNV infection is a serious threat to public health, especially to the immunocompromised and elderly22. It has also been associated with human encephalitis cases23. Two classical antiviral compounds interferon and ribavirin showed promising results in vitro24 but is unclear if these compounds are effective in patients2526. Several techniques for WNV detection, e.g. immunofluorescence technology, plaque assay and virus isolation, have been reported, but the complexity of these techniques has replaced them by more convenient methods, e.g. RT-PCR and ELISA. Early and specific detection is critical in the management of WN patients. Unless a non-infectious recombinant antigen is available, ELISA diagnosis by WNV involves working with infectious virus to produce inactivated antigens from infected cell cultures or suckling mouse brains15272829. This procedure involves risks to laboratory personnel and highly sophisticated BSL-3 containment facilities are needed to grow this zoonotic agent. Hence specific protein produced by recombinant DNA technology is being used for WNV diagnostic purpose instead of whole live virus. The present study was based on the recombinant CprM protein of WNV as a diagnostic intermediate. E. coli is the most attractive choice for recombinant protein production due to low cost of production, well-characterized genetics and cultivation conditions30. PRNT was used for the detection of neutralizing antibodies against viruses irrespective of Ig classes. PRNT was tedious and time-consuming because it takes week to complete the assay.
In conclusion, an indirect microplate ELISA using rWNV-CprM protein was standardized to detect anti-WNV IgM antibodies and evaluated with a panel of 170 suspected clinical samples. The rWNV-CprM IgM ELISA reported in this study appears to be a promising economical assay for surveillance and clinical diagnosis of WNV infection in endemic areas of developing countries.
Acknowledgment
Authors thank Dr Lokendra Singh, Director, Defence Research and Development Establishment, Ministry of Defence, Government of India, for his support, constant inspiration and providing the necessary facilities for this study. The authors also thank Dr Sivakumar Rathinam (Department of Microbiology, Aravind Eye Hospital, Madurai, Tamil Nadu, India) for providing the clinical samples.
Financial support & sponsorship: The study was funded by the Ministry of Defence, New Delhi (grant no. BTM-203 TD).
Conflicts of Interest: None.
References
- West Nile virus transmission and ecology in birds. Ann N Y Acad Sci. 2001;951:54-7.
- [Google Scholar]
- Protective and therapeutic capacity of human single-chain Fv-Fc fusion proteins against West Nile virus. J Virol. 2005;79:14606-13.
- [Google Scholar]
- Flaviviridae: The viruses and their replication. In: Fields BN, Knipe DM, Howley PM, eds. Fields virology (3rd ed). Philadelphia, PA: Lippincott-Raven; 1996. p. :931-59.
- [Google Scholar]
- Evidence for the existence of a pseudoknot structure at the 3’ terminus of the flavivirus genomic RNA. Biochemistry. 1996;35:4222-30.
- [Google Scholar]
- West Nile virus core protein; tetramer structure and ribbon formation. Structure. 2004;12:1157-63.
- [Google Scholar]
- Solution structure of dengue virus capsid protein reveals another fold. Proc Natl Acad Sci U S A. 2004;101:3414-9.
- [Google Scholar]
- Solution structure of dengue virus capsid protein reveals another fold. Biochem Biophys Res Commun. 2005;329:246-55.
- [Google Scholar]
- Isolation of capsid protein dimers from the tick-borne encephalitis flavivirus and in vitro assembly of capsid-like particles. J Virol. 2004;78:8078-84.
- [Google Scholar]
- The virology, epidemiology, and clinical impact of West Nile virus: A decade of advancements in research since its introduction into the Western Hemisphere. Epidemiol Infect. 2011;139:807-17.
- [Google Scholar]
- Flavivirus genome organization, expression, and replication. Annu Rev Microbiol. 1990;44:649-88.
- [Google Scholar]
- Rapid detection of West Nile virus from human clinical specimens, field-collected mosquitoes, and avian samples by a TaqMan reverse transcriptase-PCR assay. J Clin Microbiol. 2000;38:4066-71.
- [Google Scholar]
- Real-time reverse transcription loop-mediated isothermal amplification for rapid detection of West Nile virus. J Clin Microbiol. 2004;42:257-63.
- [Google Scholar]
- Evaluation of immunoglobulin M (IgM) and IgG enzyme immunoassays in serologic diagnosis of West Nile virus infection. J Clin Microbiol. 2000;38:2232-9.
- [Google Scholar]
- Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680-5.
- [Google Scholar]
- Genetic approach to facilitate purification of recombinant proteins with a novel metal chelate adsorbent. Biotechnology. 1988;6:1321-5.
- [Google Scholar]
- Purification of recombinant proteins with metal chelate adsorbent. Genet Eng (N Y). 1990;12:87-98.
- [Google Scholar]
- Protein measurement using bicinchoninic acid: Elimination of interfering substances. Anal Biochem. 1989;180:136-9.
- [Google Scholar]
- Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350-4.
- [Google Scholar]
- A plaque reduction test for dengue virus neutralizing antibodies. J Immunol. 1967;99:285-90.
- [Google Scholar]
- Isolation of West Nile virus from the brains of children who had died of encephalitis. Bull World Health Organ. 1984;62:879-82.
- [Google Scholar]
- Efficacy of interferon alpha-2b and ribavirin against West Nile virus in vitro . Emerg Infect Dis. 2002;8:107-8.
- [Google Scholar]
- Failure of interferon alpha-2b in a patient with West Nile virus meningoencephalitis and acute flaccid paralysis. Scand J Infect Dis. 2005;37:944-6.
- [Google Scholar]
- Clinical findings of West Nile virus infection in hospitalized patients, New York and New Jersey, 2000. Emerg Infect Dis. 2001;7:654-8.
- [Google Scholar]
- Epitope-blocking enzyme-linked immunosorbent assays for the detection of serum antibodies to West Nile virus in multiple avian species. J Clin Microbiol. 2003;41:1041-7.
- [Google Scholar]
- Genetic and phenotypic variation of West Nile virus in New York, 2000-2003. Am J Trop Med Hyg. 2004;71:493-500.
- [Google Scholar]
- Standardization of immunoglobulin M capture enzyme-linked immunosorbent assays for routine diagnosis of arboviral infections. J Clin Microbiol. 2000;38:1823-6.
- [Google Scholar]
- Enhancement of human gamma-interferon production in recombinant E. coli using batch cultivation. Appl Biochem Biotechnol. 2010;160:2366-76.
- [Google Scholar]




