
Translate this page into:
Subtelomeric rearrangements in Indian children with idiopathic intellectual disability/developmental delay: Frequency estimation & clinical correlation using fluorescence in situ hybridization (FISH)
Reprint requests: Dr Solomon F.D. Paul, Department of Human Genetics, Sri Ramachandra University, Porur, Chennai 600 116, Tamil Nadu, India e-mail: wise_soly@yahoo.com
-
Received: ,
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background & objectives:
Subtelomeres are prone to deleterious rearrangements owing to their proximity to unique sequences on the one end and telomeric repetitive sequences, which increase their tendency to recombine, on the other end. These subtelomeric rearrangements resulting in segmental aneusomy are reported to contribute to the aetiology of idiopathic intellectual disability/developmental delay (ID/DD). We undertook this study to estimate the frequency of subtelomeric rearrangements in children with ID/DD.
Methods:
One hundred and twenty seven children with idiopathic ID/DD were tested for subtelomeric rearrangements using karyotyping and FISH. Blood samples were cultured, harvested, fixed and GTG-banded using the standard protocols.
Results:
Rearrangements involving the subtelomeres were observed in 7.8 per cent of the tested samples. Detection of rearrangements visible at the resolution of the karyotype constituted 2.3 per cent, while those rearrangements detected only with FISH constituted 5.5 per cent. Five deletions and five unbalanced translocations were detected. Analysis of parental samples wherever possible was informative regarding the inheritance of the rearrangement.
Interpretation & conclusions:
The frequency of subtelomeric rearrangements observed in this study was within the reported range of 0-35 per cent. All abnormal genotypes were clinically correlated. Further analysis with array technologies presents a future prospect. Our results suggest the need to test individuals with ID/DD for subtelomeric rearrangements using sensitive methods such as FISH.
Keywords
Chromosomal
cytogenetics
developmental delay
fluorescence in situ hybridization
subtelomeric rearrangements

Intellectual disability (ID) is prevalent in 2-3 per cent of the population worldwide. While the aetiology for ID is well-established in about half of the affected individuals, it remains idiopathic in the rest1. Of the various aetiological factors, genetic causes are reported in a significant proportion; about 9.5 per cent of the affected individuals have microscopically visible chromosomal abnormalities2. The frequency of subtelomeric chromosomal rearrangements in ID averages around six per cent3; however, the range of frequencies reported varies between 0 and 35 per cent456789. It is suggested that the large variation in the reported frequencies of subtelomeric alterations may be attributed to factors such as patient selection criteria, sample size, ethnicity, method of detection, polymorphic variants and chance10.
The similarity of telomeric repetitive sequences among non-homologous chromosomes increases the probability of a recombination event. Such an event may involve the proximal neurodevelopmental genes in a deleterious rearrangement, causing ID and associated developmental delay (DD)11. Thus, the subtelomeric regions are likely candidates to test in an affected individual. The acquisition of genotype data from large population screens has become simple with the advent of novel molecular cytogenetic methods; however, the overlap in the clinical features of individuals with subtelomeric rearrangements makes the diagnosis uncertain. Hence, to characterize newer syndromes associated with ID, a definitive genotype–phenotype correlation is necessary. Therefore, in this study, we aimed to estimate the frequency of sub-microscopic rearrangements in the subtelomeric regions in children with idiopathic ID/DD using fluorescence in situ hybridization (FISH) and to find a relation between the genotype and phenotype.
Material & Methods
The experimental study was conducted between February 2012 and January 2013. One hundred and twenty seven children (7 months to 16 yr old) were enrolled for this study from August 2011 to September 2012 from the paediatric out-patient clinics (n = 92) of Sri Ramachandra University, Porur, Amrita Institute of Medical Sciences, Kochi, and Kanchi Kamakoti Childs Trust Hospital, Chennai and schools for special children (n = 35), namely, Vidya Sudha, Vasantham and Madhuram Narayanan Centre for Exceptional Children, Chennai, Tamil Naidu, India, after obtaining informed consent from the parent/caregiver. These children were recruited based on the clinical evaluation of ID/DD with at least one of the following criteria: (i) facial (>2) and/or extra-facial (>1) dysmorphism (such as, but not limited to, microcephaly, hypertelorism, clinodactyly and hypospadias); (ii) pre and/or post-natal developmental defects (such as, but not limited to, seizures, feeding difficulties and speech delay); and (iii) a positive family history for unexplained ID/related conditions (a relative with ID/DD or seizures).
Patients with a clinical history suggestive of a common aetiology or with aneuploidies detected on karyotyping were excluded from the study. A brief description of the patients is provided in Table I. The study was approved by the Institutional Ethics Committee. All experimental work was carried out in the department of Human Genetics, Sri Ramachandra University.
Metaphase chromosome analysis: Lymphocytes separated from peripheral blood (2 ml) were cultured, harvested and fixed using standardized protocols described previously12. Metaphase chromosome preparations were G-banded using trypsin, and a minimum resolution of 550 bphs was considered for analysis. Karyotypes were re-analysed retrospectively to elucidate the rearrangements detected with FISH.
Fluorescence in situ hybridization: FISH was performed for all the 127 samples, as per the manufacturer's instruction with slight modifications for standardization, on chromosome preparations using the VysisToTelVysion™ Multi-Color DNA FISH probe mixtures (Abbott Laboratories, USA) targeting the subtelomeric regions of all chromosomes, except the acrocentric ‘p’ arms. At least three slides were used for each sample, with 15 marked regions for each of the 15 probe mixes. The probes were co-denatured with target metaphase chromosomes and hybridized overnight using Hybrite™ (Abbott Vysis, USA). Excess unbound probes were removed by post-hybridization washing, and slides were counterstained with DAPI (4′,6-diamidino-2-phenylindole). Quality of the FISH experiment was evaluated based on mitotic index, chromosome morphology and signal intensities. Analysis was considered conclusive if the first 10 metaphases were concordant. In cases of discordance, the number of metaphases analysed was increased to 20. FISH was repeated for mixes showing the absence or excess of signals or indicating translocations, and the analyses were compared between two cytogeneticists. The images were documented using the CytoVision software, version 3.1 (Leica Biosystems, Germany). Where available, parental samples were also tested using FISH and GTG banding to determine whether the abnormality was inherited.
Results
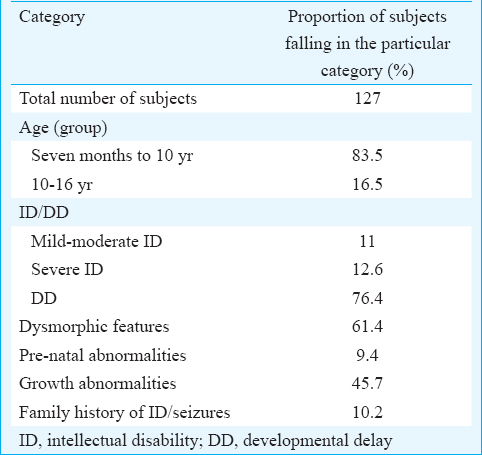
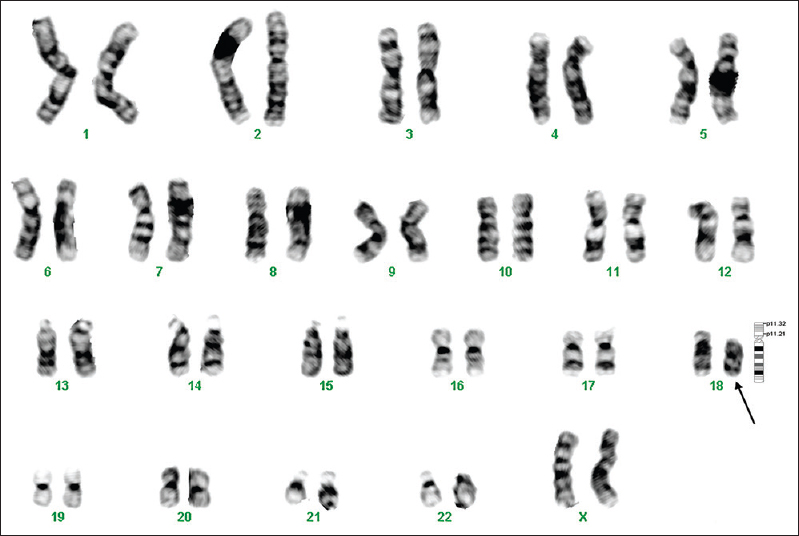
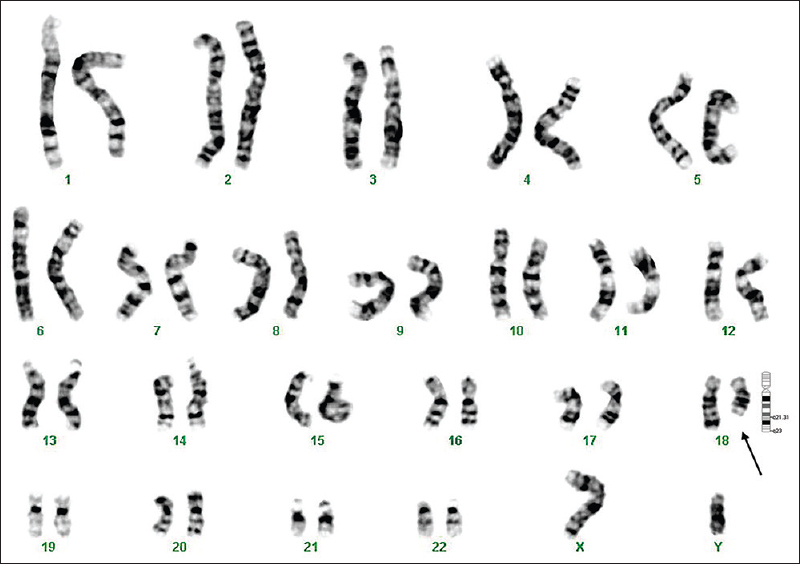
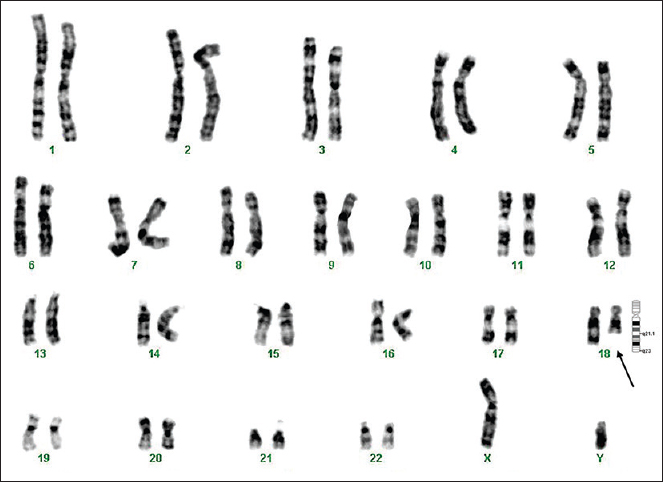
The metaphase chromosomes were successfully prepared from all blood samples collected (n = 127); analyses of G-banded metaphases showed a karyotype of 46,XY (n = 90) for normal males and 46,XX (n = 33) for normal females. Of the 127 children, 123 (97%) showed a normal karyotype and the remaining four showed an abnormal karyotype. Three abnormalities involved a deletion on chromosome 18 (patients 1, 7 and 8, Figs 1–3) and one involved an inversion on chromosome 17 (patient 6, Fig. 4).
- GTG-banded karyotype image of patient 1. The arrow points to the deletion on the ‘p’ arm of chromosome 18.
- GTG-banded karyotype image of patient 7. The arrow points to the deletion on the ‘q’ arm of chromosome 18.
- GTG-banded karyotype image of patient 8. The arrow points to the deletion on the ‘q’ arm of chromosome 18.
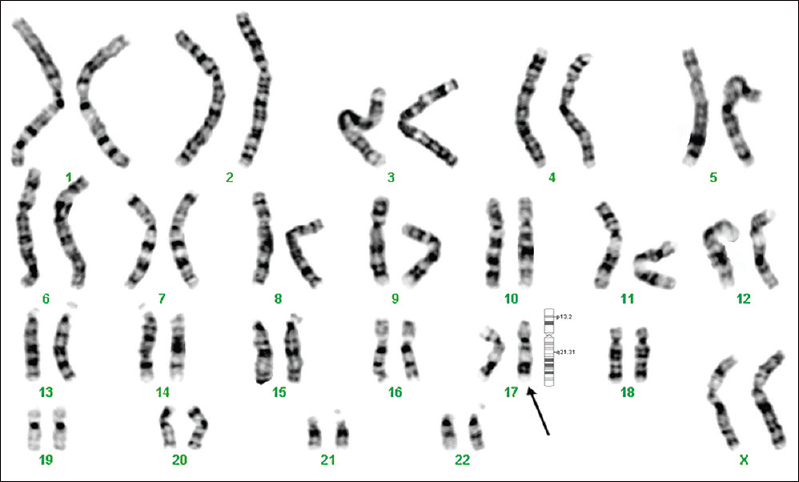
- GTG-banded karyotype image of patient 6. The arrow points to the pericentric inversion on chromosome 17.
FISH was performed to detect the subtelomeric alterations in all the patients. FISH revealed sub-microscopic rearrangements in 5.5 per cent (n = 7) and confirmed terminal deletions in those (2.3%) showing an abnormal result on GTG banding (n = 3). The overall results showed that while the frequency of abnormalities using GTG banding alone was 2.3 per cent, FISH as a stand-alone technique yielded a value of 7.8 per cent.
The clinical description and chromosomal abnormalities of the patients with subtelomeric rearrangements are described in Table II. Overall, five children showed deletions and five showed an unbalanced translocation, probably inherited from a parent who was a balanced translocation carrier. The deletions observed were 18p-, 18q- (patient 7, Fig. 5) and 17p- (patient 6, Fig. 6) and 4p-. The unbalanced translocations were 7q-3q+ (patient 2, Fig. 7a and b), Yq-Yp+, 1q-5p+, 4p-8p+, 6p-8p+. Parental samples were available for four children, two of whom showed an apparently balanced translocation: mother of patient 2 showed a balanced translocation between 3q and 7q (Fig. 8a and b) and the father of patient 9 showed a balanced translocation between 4p and 8p. The parents of the other two patients showed no chromosomal abnormality, indicating a de novo deletion in the child. The karyotype and FISH results are presented in Table III. In addition, subtle deletions in two patients were apparent on re-analysis of the karyotypes, with the knowledge of the FISH results.

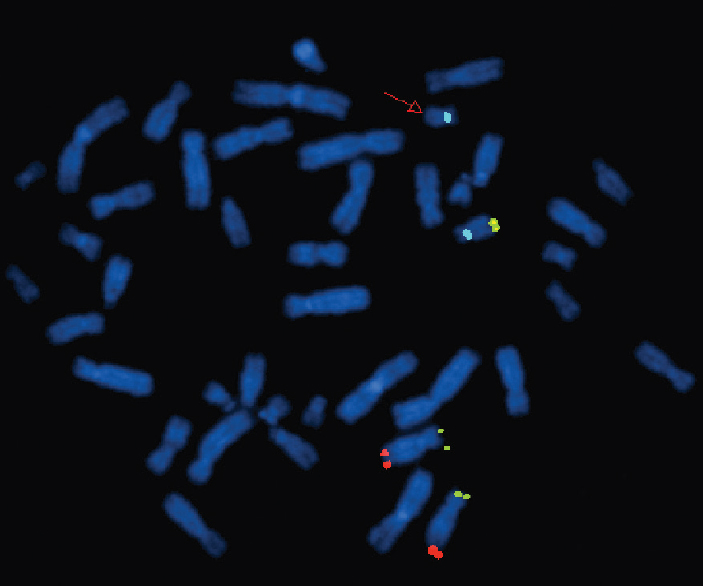
- Fluorescence in situ hybridization image of metaphase plates of patient 7 - Mix 12 with probes for telomeres of chromosome 12p (green), 12q (red), 18q (yellow) and control region, centromere 18 (aqua). The red arrow points to the absence of yellow signal on one of the chromosome 18s, indicating a deletion of 18qter.
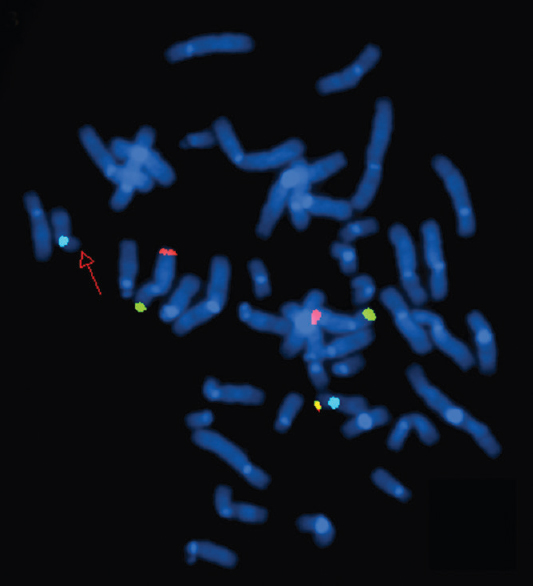
- Fluorescence in situ hybridization image of metaphase plates of patient 6 - Mix 8 with probes for telomeres of chromosome 8p (green), 8q (red), 17p (yellow) and control region, centromere 17 (aqua). The red arrow points to the absence of yellow signal on one of the chromosome 17s, indicating a deletion of 17pter.
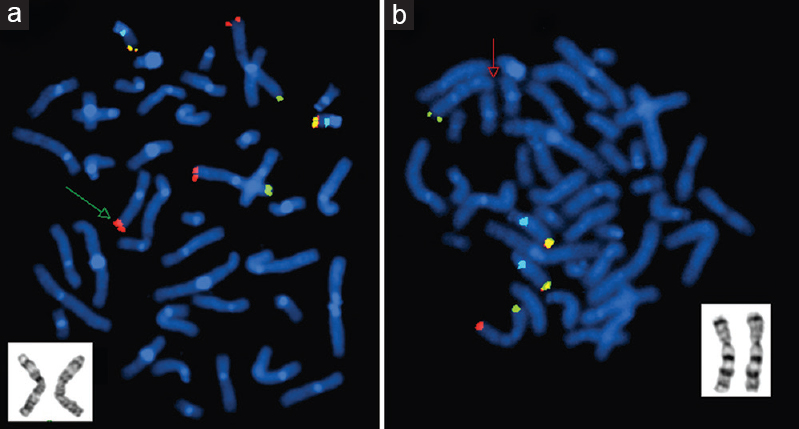
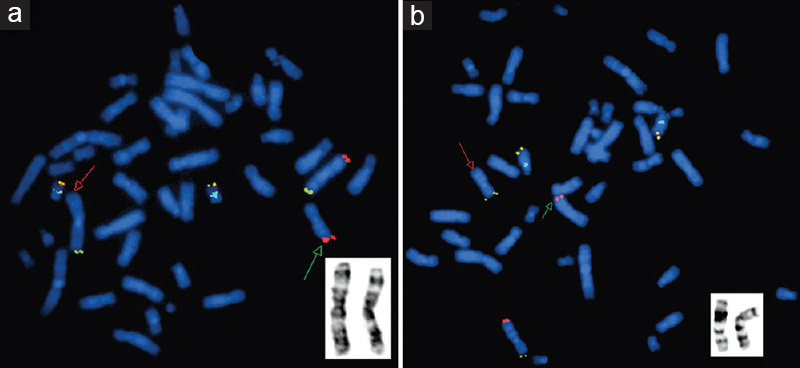
- Fluorescence in situ hybridization image of metaphase plates of patient 2 - (a) Mix 3 with probes for telomeres of chromosome 3p (green), 3q (red), and 22q (yellow) and control region, 22q11 (aqua). The green arrow points to the presence of an additional red signal on one of the chromosome 7s, indicating a derivative 7 with duplicated 3qter. GTG-banded partial karyotype in the inset shows both homologues of chromosome 3. (b) Mix 7 with probes for telomeres of chromosome 7p (green), 7q (red), and 14q (yellow) and control region, 14q11.2 (aqua). The red arrow points to the absence of red signal on one of the chromosome 7s, indicating a deletion of 7qter. GTG-banded partial karyotype in the inset shows both homologues of chromosome 7.
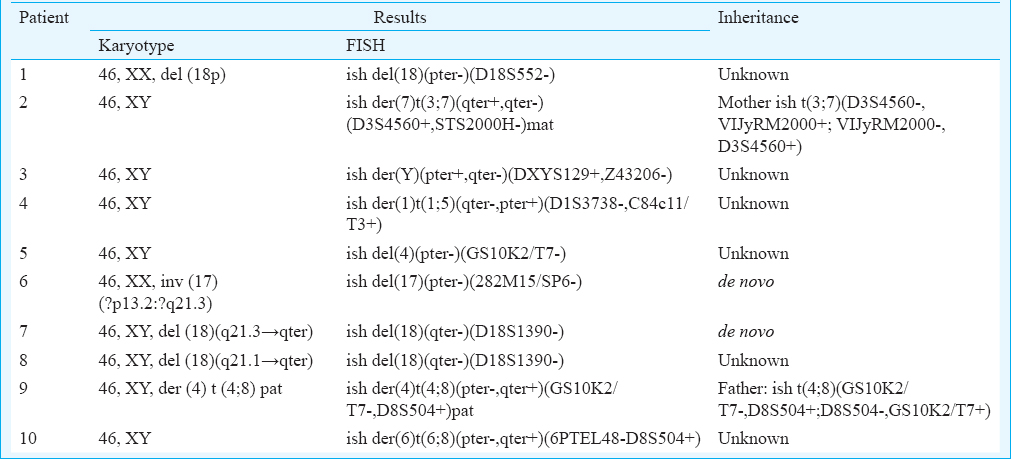
- Fluorescence in situ hybridization image of metaphase plates of the mother of patient 2 - (a) Mix 3 with probes for telomeres of chromosome 3p (green), 3q (red), and 22q (yellow) and control region, 22q11 (aqua). The red arrow points to the absence of red signal on chromosome 3qter, and green arrow points to the presence of red signal on chromosome 7qter, indicating t(3;7). GTG-banded partial karyotype in the inset shows both homologues of chromosome 3. (b) Mix 7 with probes for telomeres of chromosome 7p (green), 7q (red), and 14q (yellow) and control region, 14q11.2 (aqua). The red arrow points to the absence of red signal on chromosome 7qter, and the green arrow points to the presence of red signal on chromosome 3qter, indicating an apparently balanced reciprocal t(3;7). GTG-banded partial karyotype in the inset shows both homologues of chromosome 7.
Discussion
The subtelomeric regions are recombination hotspots, whose rearrangements may lead to segmental aneusomy and haploinsufficiency of genes in the flanking regions. With at least one-third of the total human genes being expressed in the brain13, the probability of a subtelomeric rearrangement resulting in a neurodevelopmental disorder is extremely high. Continual research in this area has led to the characterization of distinct syndromes associated with subtelomeric rearrangements.
The abnormalities described in our study included 18p-, 7q-3q+, Yq-Yp+, 1q-5p+, 4p-, 17p-, 18q-, 4p-8p+ and 6p-8p+. Some of these rearrangements have been reported previously; however, the phenotypes and levels of severity are variable. About 10 per cent of cases with 18p- have been reported to have brain malformations as part of the holoprosencephaly spectrum, and in general, the deletions reported on 18p are microscopically visible14. 3q29 duplication and 7q36 deletion syndromes have been reported either as the sole abnormality or with the involvement of another chromosome. A three generation family in which five members had mild-to-moderate ID and minor dysmorphic features associated with an interstitial micro-duplication of chromosome 3q29 has been reported15. The 7q36.1-qter region has been reported to comprise two genes: the HLXB9 gene involved with Currarino syndrome (sacral dysgenesis, anorectal atresia and a presacral mass) and the sonic hedgehog gene involved with holoprosencephaly1617. The SHOX gene on Yp11.3 is reported to be involved in growth retardation and cause short stature, however, may not affect intelligence18. Joyce et al had reported a complex chromosomal rearrangement with nine breakpoints involving five chromosomes including the Y chromosome in a boy with mild ID, DD, short stature and microcephaly19. The deletion on 1q44 is thought to encompass multiple genes in the region, and thus, the manifestations are a result of the contiguous gene deletion and no specific genes have been implicated. Duplication of 5p-p14.3 has been reported in two sisters with ID, obesity, mandibular prognathism with eye and skin anomalies2021. The Wolf-Hirschhorn syndrome (WHS) critical region is located in 4p16.3. Deletions with a size <3.5 Mb have been described in the mild variant of WHS, also known as Pitt-Rogers-Danks syndrome, with the critical deletion region narrowed down to 165 kb at 4p16.3. The loss of multiple genes on 4p such as WHSC1, LETM1 and MSX1, which are thought to play significant roles in early development, is reported to cause the typical features of the syndrome14. Terminal deletions of 17p have been known to result in a distinctive phenotype, Miller-Dieker syndrome, a contiguous gene syndrome. However, phenotypically normal subjects with telomeric 17p deletions up to 600 kb have also been reported2223. Since the patient in our study did not show classical features of Miller-Dieker syndrome or isolated lissencephaly, the clinical significance of this de novo deletion was indeterminate. The breakpoints as well as clinical presentation for 18q deletion syndrome vary greatly among reports, suggesting that there are no hotspots. However, various critical regions have been identified, including a 4.3-Mb region within 18q22.3-q23 that is responsible for the typical 18q deletion phenotype24. Monosomy 4p with trisomy 8p results in a WHS phenotype. However, since the extent of the deletion is minimal, the features are atypical. The deletion of region 6p up to 6p24 has been implicated in various syndromes. Patients with 6p deletion were characterized with DD and hypotonia, with an abnormal skull shape25. The significance of the presence of three copies of 8p23, however, is not known. Inverted duplications of the region proximal to 8p23.3, without involvement of the subtelomeric region, have been reported with clinical abnormalities26. The significance of trisomy 8p in our patients remained unclear; the loss of gene-rich regions on 4p and 6p as well as the excess of genes on 8p could be responsible for the observed phenotype.
The frequency of subtelomeric rearrangements in individuals with ID ranges from 0 to 35 per cent456789. The lack of concordance on the reported frequencies among studies may be due to various factors. Patient inclusion criteria vary among studies and that is known to influence the observed rearrangement frequency. The largest study so far on 11688 subjects with ID reported an abnormality rate of only three per cent5. The low frequency observed in their study was attributed to the study population being unselected. Our study employed less stringent inclusion criteria that did not adhere to the de Vries et al checklist27. However, the frequency observed in our study was within the range of published reports.
The presence of polymorphic variants among subtelomeric rearrangements has been reported in healthy control groups or in the families of affected subjects in a few studies45. It has also been reported that the subtelomeres of certain chromosomes such as 2q and 10q may be polymorphic528. Ravnan et al5 described partial deletions of the 10q telomere as deduced by their diminished fluorescent intensity in 14 subjects as variants, some of them familial. In our experience, FISH analysis of metaphases from eight patients showed reduced fluorescence intensity in one of the 10q telomeres. However, since the test is not quantitative and as parental samples for these subjects were unavailable, it remained unsubstantiated to classify them as polymorphic variants.
It is not known whether subtelomeric rearrangement frequencies differ among various ethnic groups. Our study reports the frequency in a sample of the Indian population, from which data are limited. A previous report from India did not detect any subtelomeric rearrangement using multiplex ligation-dependant probe amplification in a study population of 122 individuals1.
The association of the phenotypes with the deduced genotypes depends on further characterization of the regions involved in the rearrangements at higher resolution. An assessment of the whole genome may be the best possible approach; however, it is not feasible in every case. Technologies such as array CGH have revolutionised the field of molecular cytogenetics, however, these still require standardization, validation and cost optimization.
In conclusion, our study reiterates the necessity to screen patients with unexplained ID/DD for subtelomeric rearrangements, which may be missed in routine cytogenetic testing, using a sensitive technique such as FISH. When the clinical presentation warrants a genetic diagnosis, screening the genomic hotspots at high resolution is recommended.
Acknowledgment
Authors acknowledge the Indian Council of Medical Research, New Delhi, India, for financial support, and thank Ms. Raheema Beevi for assistance. Authors also thank the participants from Vidya Sudha Special School, Vasantham Special School and Madhuram Narayan Centre for Exceptional Children, Chennai, Tamil Nadu.
Conflicts of Interest: None.
References
- Multiplex ligation-dependant probe amplification study of children with idiopathic mental retardation in South India. Indian J Hum Genet. 2013;19:165-70.
- [Google Scholar]
- Diagnostic investigations in individuals with mental retardation: A systematic literature review of their usefulness. Eur J Hum Genet. 2005;13:6-25.
- [Google Scholar]
- Subtelomeric rearrangements: Results from a study of selected and unselected probands with idiopathic mental retardation and control individuals by using high-resolution G-banding and FISH. Hum Genet. 2001;109:440-51.
- [Google Scholar]
- Subtelomere FISH analysis of 11 688 cases: An evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J Med Genet. 2006;43:478-89.
- [Google Scholar]
- Subtelomeric chromosomal rearrangements in a large cohort of unexplained intellectually disabled individuals in Indonesia: A clinical and molecular study. Indian J Hum Genet. 2013;19:171-8.
- [Google Scholar]
- Screening for submicroscopic chromosome rearrangements in children with idiopathic mental retardation using microsatellite markers for the chromosome telomeres. J Med Genet. 1999;36:405-11.
- [Google Scholar]
- Subtle chromosomal rearrangements in children with unexplained mental retardation. Lancet. 1999;354:1676-81.
- [Google Scholar]
- Comparative genomic hybridisation in mentally retarded patients with dysmorphic features and a normal karyotype. Clin Genet. 2001;60:212-9.
- [Google Scholar]
- Prospective screening for subtelomeric rearrangements in children with mental retardation of unknown aetiology: the Amsterdam experience. J Med Genet. 2002;39:546-53.
- [Google Scholar]
- Perfect endings: A review of subtelomeric probes and their use in clinical diagnosis. J Med Genet. 2000;37:401-9.
- [Google Scholar]
- Chromosome preparations of leukocytes cultured from human peripheral blood. Exp Cell Res. 1960;20:613-6.
- [Google Scholar]
- Genetic influences on brain developmental trajectories on neuroimaging studies: from infancy to young adulthood. Brain Imaging Behav. 2014;8:234-50.
- [Google Scholar]
- 3q29 interstitial microduplication: A new syndrome in a three-generation family. Am J Med Genet A. 2008;146A:601-9.
- [Google Scholar]
- A homeobox gene, HLXB9, is the major locus for dominantly inherited sacral agenesis. Nat Genet. 1998;20:358-61.
- [Google Scholar]
- Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357-60.
- [Google Scholar]
- Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet. 1997;16:54-63.
- [Google Scholar]
- A de novo complex chromosomal rearrangement with nine breakpoints characterized by FISH in a boy with mild mental retardation, developmental delay, short stature and microcephaly. Clin Genet. 1999;56:86-92.
- [Google Scholar]
- Mental retardation, obesity, mandibular prognathism with eye and skin anomalies (MOMES syndrome): A newly recognized autosomal recessive syndrome. Am J Med Genet. 2001;103:283-8.
- [Google Scholar]
- Obesity syndrome, MOMES caused by deletion-duplication (4q35.1 del and 5p14.3 dup) Am J Med Genet A. 2009;149A:833-4.
- [Google Scholar]
- “Molecular rulers” for calibrating phenotypic effects of telomere imbalance. J Med Genet. 2002;39:734-40.
- [Google Scholar]
- Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am J Hum Genet. 2003;72:918-30.
- [Google Scholar]
- Genotype-phenotype mapping of chromosome 18q deletions by high-resolution array CGH: An update of the phenotypic map. Am J Med Genet A. 2007;143A:1858-67.
- [Google Scholar]
- Clinically abnormal case with paternally derived partial trisomy 8p23.3 to 8p12 including maternal isodisomy of 8p23.3: A case report. Mol Cytogenet. 2009;2:14.
- [Google Scholar]
- Clinical studies on submicroscopic subtelomeric rearrangements: A checklist. J Med Genet. 2001;38:145-50.
- [Google Scholar]
- Subtelomeric rearrangements in idiopathic mental retardation. Indian J Pediatr. 2005;72:679-85.
- [Google Scholar]




