
Translate this page into:
Microbiome: Paediatricians’ perspective
Reprint requests: Dr Shilpa Khanna Arora, Department of Pediatrics, Postgraduate Institute of Medical Education & Research & Dr Ram Manohar Lohia Hospital, New Delhi 110 001, India e-mail: drshilpakhanna@yahoo.co.in
-
Received: ,
This is an open access article distributed under the terms of the Creative Commons Attribution NonCommercial ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Millions of microorganisms inhabit the human body and affect its homeostasis in multiple ways. Alterations in this microbial community have implications for the health and survival of the human hosts. It is believed that these microorganisms should be included as part of the human genome because of their influence on human physiology hence the term “microbiome” is commonly used to refer to these microbes along with their genetic make-up and their environmental interactions. In this article we attempt to provide an insight into this recently discovered vital organ of the human body which is yet to be fully explored. We herein discuss the composition and role of microbiome in human health and disease with a special emphasis in children and culture-independent techniques employed in mapping of the microbiome. Alteration in the gut microbiome has been associated with causation of several paediatric diseases like infantile colic, necrotizing enterocolitis, asthma, atopy, obesity, type -1 diabetes, and autism. Atopic dermatitis and psoriasis have also been associated with changes in the cutaneous microbiome. Respiratory microbial imbalances during infancy have been linked with wheezing and bronchial asthma. Dysbiosis in the regional microbiome has been linked with caries, periodontitis, and chronic rhinosinusitis. The future therapeutic implications of this rapidly evolving area of research are also highlighted.
Keywords
Children
metagenomics
microbiome
microbiota
16S RNA
virome

Introduction
Humans inhabit a whole community of microorganisms in and on their body1. Almost 100 trillion symbiotic microbes inhabit a single human body which is almost 10 times the number of cells present in an adult human. It amounts to almost 1-3 per cent of the body weight2. The term “microbiome” was first used by Joshua Lederberg3 for “the ecological community of commensal, symbiotic, and pathogenic microorganisms that literally share the human body”. He suggested that these microorganisms should be included as part of the human genome because of their influence on human physiology. Research in the field of metagenomics suggests that the brigade of microorganisms living in close proximity, both in and on, human beings is indispensible for human survival. This has led to the hypothesis that the human being must be regarded as a super-organism in whom symbiotic microorganisms perform multiple tasks ranging from digestion of food to angiogenesis45. The disruption in this flora is associated with many disease conditions ranging from diarrhoea to neoplasia.
Basic terminology
The more commonly used term ‘microflora’ or microbial ‘flora’ is actually a misnomer as it technically means the plant or vegetable microbial community of a region. Recently, the term ‘micobiota’ has become prevalent which literally means all the living organisms of a region. Considering the fact that these organisms are an integral part of the human genetic landscape, the term ‘microbiome’ seems to be best suited as it symbolises all the microorganisms along with their genetic make-up as well as their interactions in a particular environment3. Conventionally, the major part of the research relevant to human microbiome has focussed on identification and study of the inhabiting bacteria, however, there also exists a ‘virome’ i.e. the viruses inhabiting the body, that are probably as important6. ‘Phylotypes’ or ‘operational taxonomic units’ (OTUs) are a group of microbes generally defined by the level of sequence similarity (percentage) between the 16S rRNA genes (e.g. ≥ 98% for a ‘species’-level phylotype)7.
The recent advent in the knowledge of human microbiome is undoubtedly attributed to the preceding advances in the field of metagenomics. ‘Metagenomics’ is the science of directly analysing the genome (the complete set of DNA present in a single cell) contained in an environmental sample8. Metagenomics has opened up the avenues for obtaining the genetic information on potential biocatalysts, genomic links between function and phylogeny along with the evolutionary profile of the microbial community8. Three closely related fields that are coming up are ‘metatranscriptomics’ i.e. the study of the transcriptomes (reverse-transcribed RNA transcripts) of a group of interacting microbes9, ‘metaproteomics’ i.e. the study of the entire protein complement of the microbial community10 and ‘metabolomics’ i.e. the study of small-molecule metabolites of the microorganisms11.
Studies carried out in ‘germ-free animals’ i.e., the ones born and reared in sterile environments free of any microbiota have provided useful insight into the complex interactions taking place between the microbiome and its host12. Likewise, ‘gnotobiotic systems’ have been developed to get animals with desired microbiome by transferring or synthetically implanting the desired microbes from another host into the germ-free animals13. Researchers have developed gnotobiotic systems with humanized microbiota to evaluate the effects of controlled interventions14.
Mapping the microbiome
The foundation of present day microbiology was laid down in the nineteenth century after Robert Koch developed the technique of selectively growing and isolating bacteria in culture medium. Until recently we were dependent on the conventional methods of ‘cultivating’ bacterial ‘pure cultures’ for isolating the microorganisms in a sample and then depending on growth on a particular type of medium, colony morphology and consumption or production of a particular metabolite to finally identify it. But the observation that only a few of the microbes out of the many visualised microscopically could be isolated by culture prompted scientists to develop the newer ‘culture-independent’ techniques to recognise these yet unidentified microbes15. Escherichia coli that was considered as the major component of gut microbiota actually constitutes much less than 1 per cent of gut bacteria, but as it grows easily in culture, it can be detected even at low abundance16. It is estimated that almost 20-60 per cent of our microbiome, depending on the region of the body, cannot be cultured by the conventional methods17.
The prokaryotic cell contains 16S RNA gene in its ribosome that has about 1500 nucleotide; it is highly conserved between different species of bacteria but has several hypervariable regions that can allow identification at genus or species level13. Sequence analysis of 16S rRNA gene is thus utilised in many phylogenetic studies to simultaneously amplify the genetic regions of clusters of diverse bacteria by PCR. Initial DNA sequencing techniques were based on tedious fingerprinting methods for the separation of 16S RNA like denaturing gradient gel electrophoresis (DGGE) and restriction fragment length polymorphism (RFLP)12. Sanger technique which was developed in 1977 was used for more than two decades for sequencing of the amplified and cloned 16S RNA. It was based on the classical chain termination methods which could be used only on short strands (100-1000 base pairs). Cloning longer DNA strands became possible with the advent of Shot-gun sequencing by which longer sequences could be subdivided into smaller fragments, and subsequently re-assembled to give the overall sequence18. With the recent technological explosion, many low-cost, high-throughput sequencing technologies have been developed that parallelize the sequencing process and can produce millions of sequences concurrently. These ‘highly-parallelized’ or ‘next-generation’ sequencing techniques like 454 Pyrosequencing, Sequencing by Synthesis (Illumina) And Sequencing by Ligation (SOLiD) are much more accurate and less time consuming19. Whole-Genome Shotgun (WGS) metagenomic sequencing has emerged as an important strategy enabling scientists to analyze the DNA extracted directly from a sample and evaluate not only its composition (taxonomic diversity), but also the metabolic tasks (functional metagenomics) performed by the microbial community1213. The sequenced clones are utilised for the purpose of identification by finding the closest match in the existing gene database bank if available; and the novel sequences are added to the database to facilitate future research.
Virus identification is a relatively tougher task as the viruses lack this 16S RNA gene. Probably that is the reason why the researchers believe that virome research has lagged much behind than that has occurred for their bacterial counterparts6. But with the advent of shotgun sequencing that enables deciphering each and every sequence of DNA in the sample, viral metagenomics is being utilised to discover the role of human virome in health and disease2021.
The task of mapping the human microbiome has been taken up by scientists around the world through large scale projects like Human Microbiome Project (HMP) and Metagenomics of the Human Intestinal Tract (MetaHIT). The United States National Institutes of Health (NIH) launched the HMP in 2008 with the goal of characterizing healthy human microbiome using the culture-independent methods, to determine whether perturbations in the microbiome affect health/disease status, to provide a standardized data resource and new technological approaches to enable further research, and to evaluate the ethical, legal, and social implications of the same17. The European Commission initiated the MetaHIT in 2008 to decipher the intestinal metagenome and analyze their association with human phenotypes22. These projects have helped scientists to discover an enormous database comprising thousands of taxonomic profiles that constitute more than trillion bytes of data of metagenomic sequences2223.
Composition and biodiversity of healthy human microbiome
The human body is home for taxonomically diverse classes of friendly microbes ranging from eukaryotes, archaea, bacteria and even viruses23. The gastrointestinal tract is the largest reservoir of commensals in the body and hence has been studied most extensively121424252627. Other sites which have been sampled and analysed include skin, oral cavity, nasal cavity, lower respiratory tract, and vagina. Costello et al26 in their study of multiple body sites were able to detect members of 22 bacterial phyla, of which > 90 per cent was contributed by four predominant phyla, viz., Actinobacteria (36.6%), Firmicutes (34.3%), Proteobacteria (11.9%), and Bacteroidetes (9.5%)26282930. Though the number of phyla is limited, the biodiversity increases at the level of class, family, genus and becomes enormous at the level of species24. Each body habitat is dominated by certain signature microbes like Propionibacterium on the skin and Lactobacillus in the vagina173132.
Bacteroidetes and Firmicutes are the predominant phyla inhabiting the gut amounting to more than 95 per cent of the adult gut microbiome25262730. Family members share more similar gut microbiome as compared to unrelated individuals733. Each adult individual has a unique microbiome whose composition tends to remain stable over a period of time33. Neonates are born with an almost sterile gastrointestinal tract. Environmental exposures during infancy lead to dense colonization of the gut that is highly variable over time but by the end of infancy the microbiome converges to resemble almost like an adult33. The amount and variety of microorganisms inhabiting the gut during this time have a significant impact on the development of a person's immune system with life-long consequences34. Multiple factors affect the timing and composition of this ecosystem in the neonatal gut. Penders et al35 observed that infants delivered by caesarean section had lower colonization rates and counts of bifidobacteria and Bacteroides fragilis but higher prevalence and counts of Clostridium difficile and E. coli. Breastfed infants at the age of one month were predominantly colonized with bifidobacteria whereas formula-fed infants were colonized with E. coli, C. difficile, B. fragilis, and lactobacilli. Hospitalization and prematurity were associated with higher rates of colonization with C. difficile. Use of oral antibiotic decreased the levels of obligate anaerobes like bifidobacteria and Bacteroides. Infants having an older sibling demonstrated higher bifidobacterial counts. They concluded that term infants, delivered vaginally at home and exclusively breastfed have the most beneficial gut microbial composition, i.e. having large number of bifidobacteria and less of C. difficile and E. coli35. Antibiotics are known to inhibit the healthy microbiota allowing pathogenic microbes like C. difficile to proliferate. Antibiotic exposure, direct as well as indirectly through breast milk in neonates has been shown to result in significant alterations in the gut microbiome that may last for days to months3536.
The predominant phyla present on the skin are Actinobacteria, Firmicutes, and Proteobacteria whereas Bacteroidetes which is predominant in the gut is a minor component of the skin3738. In comparison to adults, infant skin shows Firmicutes predominance39. Akin to gut microbiome, the cutaneous microbiome also evolves during infancy with staphylococci dominance during the initial period giving way to a more diverse ecosystem by the end of the first year39. Skin is an indispensible physical and immune barrier for humans hence its early microbial colonization is an important determinant of body's defence against pathogens. The microbial composition of infants as well as adults is highly variable but site specific depending upon local anatomy, lipid content, pH, sweat, and sebum secretion at the site40. This could be the underlying reason as to why certain diseases of the skin that have been linked with microbial causation have predilection for specific skin sites like acne, atopic dermatitis, psoriasis and seborrhoeic dermatitis40.
Majority of the bacteria sampled at the nostrils belong to Firmicutes and Actinobacteria; but Proteobacteria are very few unlike the skin elsewhere41. Oropharynx is known to inhabit microbes belonging to Firmicutes, Proteobacteria, and Bacteroidetes41. Till date, very few researchers have been able to characterize the healthy lower respiratory tract microbiome as obtaining pure lung derived samples not contaminated by upper airway microbes is a relatively difficult task. Charlson et al42 have observed that the lung microbiome is similar in composition to upper respiratory tract but has a lesser biomass and hypothesised that it probably originates by micro-aspiration from upper airways. Pyrosequencing techniques have established that the female genital microbiome is dominated by lactobacilli that belong to phylum Firmicutes32.
It is not only the taxonomic composition, but also the taxonomic diversity that has been implicated in causation of several diseases. Two parameters that are routinely employed for this purpose are alpha diversity, i.e. how many kinds of taxa or lineages are within a sample and beta diversity, i.e. how many kinds of taxa or lineages are shared among samples from same habitat among different subjects43. Data from NIH-HMP demonstrated that saliva had the maximum variety of bacteria (highest alpha diversity) but different individuals had similar microorganisms (lowest beta diversity) in their saliva43. On the other hand, skin microbiome showed intermediate alpha diversity but highest beta diversity, whereas vaginal samples demonstrated the lowest alpha as well as beta diversity at the genus level. Temporal analysis has revealed that each adult harbours a unique microbiome that stays more or less stable over time as compared to the population as a whole both in terms of microbial composition as well as metabolic functions43.
Studies evaluating the metagenomics have proved that in spite of variations in the taxonomic profile, the metabolic functions carried out by the microbiome of a particular body site were similar among different individuals. This has prompted scientists to hypothesise about the possible existence of a ‘Core Microbiome’ that might be sharing a set of genes and/or metabolic capabilities7. Tap and colleagues30 observed that inspite of presence of a large number of species in the gut a limited number of OTUs were shared amongst many individuals and hence these might represent the phylogenetic core of human gut microbiome. Recently, Li and colleagues44 attempted to characterize the core microbiome of different body habitats using two parameters viz. ubiquity and abundance. These have been represented in the Figure.
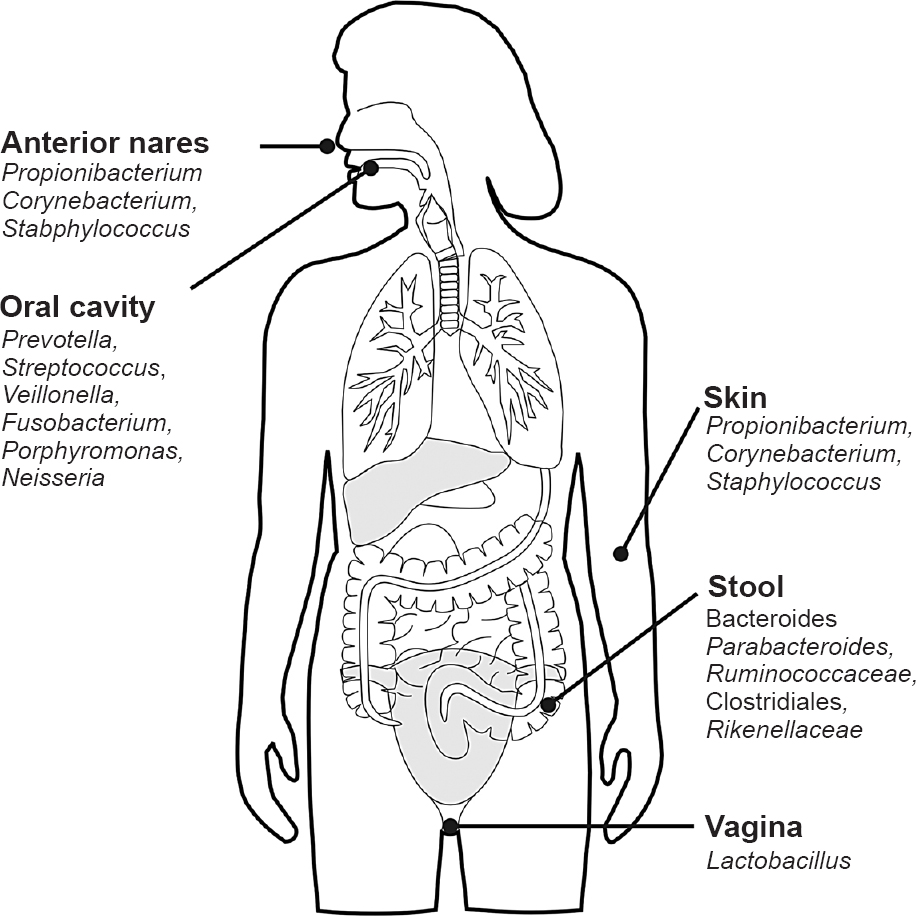
- Representation of the core taxa at different human microbial habitats.
Microbiome and diseases of childhood
Gut microbiome plays a vital role in several body functions like nutrient processing and assimilation; defence against pathogenic microbes; and even stimulation of angiogenesis454546. Alteration in this ecosystem has been associated with causation of several diseases of the gut in children, like infantile colic and necrotizing enterocolitis (NEC)4748495051. Imbalances in intestinal microbiome have been implicated in causation of many non-gastrointestinal disorders as well, like asthma, atopy, obesity, type -1 diabetes, and autism as depicted in the Table727474849505152535455565758596061626364.
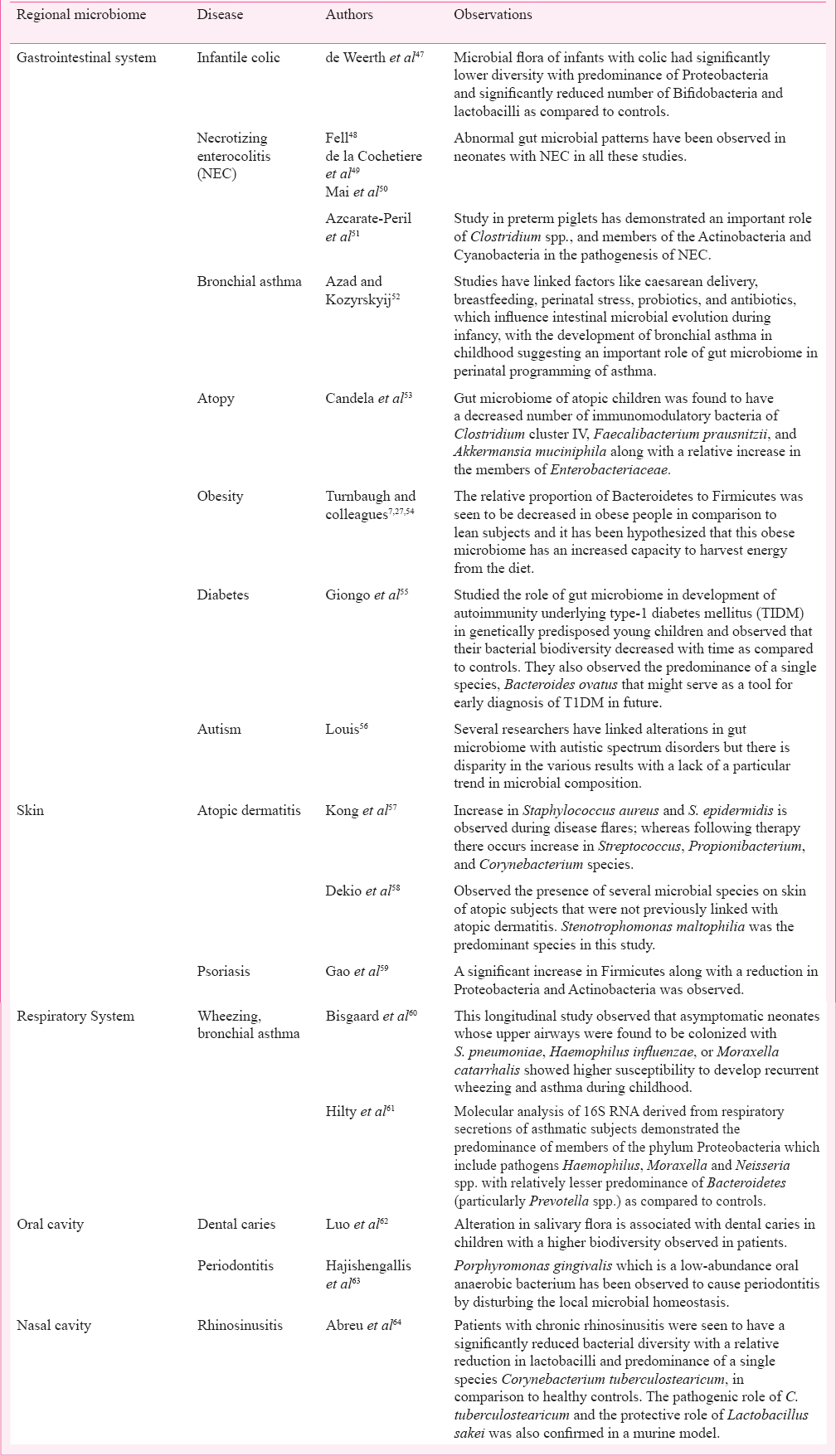
Several skin disorders like atopic dermatitis and psoriasis have been associated with changes in the local cutaneous microbiome575859. Similarly alterations in respiratory microbiome during infancy have been linked with wheezing as well as future development of bronchial asthma6061. Researchers have demonstrated the role of altered microbiome in many oronasal diseases like caries, periodontitis, and chronic rhinosinusitis626364.
Virome
Majority of the viruses that inhabit humans are the bacteriophages, i.e. viruses that infect bacteria. Hence the human virome apart from influencing the cellular processes directly could be acting indirectly by altering the symbiotic bacterial functioning, composition, or abundance6. Lysholm et al21 analysed the respiratory secretions of children with severe lower respiratory tract infection by metagenomic sequencing and observed that three of the RNA virus families were responsible for more than 90 per cent of these infections. These were Paramyxoviridae - human respiratory syncytial virus (hRSV), human metapneumovirus (hMPV) and human parainfluenza virus (hPIV); Orthomyxoviridae - influenza virus; and Picornaviridae - human rhinovirus (HRV)21. It has been observed that viruses that may be non-pathogenic otherwise, interact with various susceptibility genes in predisposed individuals and result in diseases like Crohn's disease, type-I diabetes and bronchial asthma which would not have manifested if either the susceptibility allele or the virus was absent6566. On the other hand, certain pathogenic viruses like herpes viruses tend to adapt to the host body causing lifelong infection, are at times considered a part of the human virome67. EBV and other herpes viruses have been implicated in causation of allergic and atopic diseases like asthma and eczema by immunomodulatory mechanisms67. Wylie et al68 carried out sequence analysis of the human virome in nasal swabs from febrile and afebrile children and observed that children with unexplained fevers had more viruses than healthy kids hinting towards a viral aetiology. This knowledge could be utilized in developing tests that would help in rapid identification of the virus and thus avoid unnecessary antibiotic usage.
Microbiome research in children in the Indian context
Very few paediatric studies have been carried out in India to evaluate the role of microbiome in health and disease. A study from Pune observed that the intestinal flora of infants born by caesarean section was more diverse as compared to infants delivered vaginally69. The most abundant bacterial species present in vaginally delivered infants were Acinetobacter spp., Bifidobacterium spp. and Staphylococcus spp. whereas caesarean delivered infants’ faecal microbiota was dominated by Citrobacter spp., E. coli and C. difficile but lacked in Bifidobacterium spp. In another study published from Vellore, it was observed that one species of Bifidobacterium i.e. B. longum subspecies infantis colonized and predominated the neonatal gut70. They also observed that asymptomatic rotavirus infection in neonates did not alter the development of the intestinal microbiota in terms of bifidobacterial diversity or colonization.
Future implications
The observed gut microbiome dysbiosis in diseases like gastroenteritis, necrotizing enterocolitis, inflammatory bowel disease, malabsorption, obesity and atopy can open up the avenues for prevention and management of these diseases by several ways. Animal studies have demonstrated that many gut bacteria produce immunomodulatory, anti-inflammatory, and growth promoting molecules717273. These microorganisms also produce antibacterial substances like bacteriocin and lacticin that inhibit the growth of pathogens like C. difficile7475. Further characterization of human microbiome might help to utilize these bacterial derivatives as therapeutic agents to control various disease states. The term “pharmabiotic” has thus been used to denote any material that has been obtained from the intestinal microbiome and can be utilized for health promotion, be it a molecular by- product or a microorganism itself.
Microbiological modification of gut microbiome in a desired manner can be attempted by means of administering various prebiotics and probiotics, separately or in combination as synbiotics. These “functional food ingredients” are being utilized to enrich the gut microbiome with bacteria like Bifidobacterium and Lactobacillus that are believed to be health promoting76. The role of prebiotics and probiotics has been well established in acute gastroenteritis as well as antibiotic associated diarrhoea. In a meta-analysis by Deshpande et al77, it was observed that probiotic supplementation in preterm neonates not only reduced the risk of necrotizing enterocolitis but also the risk of mortality. Similarly in a meta-analysis by Brenner et al78, probiotic usage in patients of irritable bowel syndrome has been found to be efficacious.
Faecal microbiota transplantation (FMT) has been tried by occasional researchers in the last century also as a therapeutic intervention for antibiotic associated diarrhoea caused by C. difficile. A systematic review found out its efficacy to the tune of 92 per cent but further research is required to standardize the procedure; evaluate its safety and suitability before it is approved as a treatment modality by the regulatory agencies7980.
de Weerth et al47 in their study on infants with colic, observed the consistent presence of a few Proteobacteria linked to Escherichia, Klebsiella, Serratia, Vibrio, Yersinia, and Pseudomonas with the colic phenotype. Saulnier et al81 have been able to link specific “signature” microbes with several diseases like irritable bowel syndrome. These disease specific signature phylotypes might serve to devise diagnostic as well as therapeutic strategies in the future.
Recent advances in pharmacogenomics that focus on the role of genetics in an individual's response to a drug have prompted researchers to explore other environmental influences that affect drug metabolism. Gut microbiome is one such important determinant. Personalized drug therapy to improve efficacy and reduce adverse effects might become feasible with an approach utilizing pre-dose metabolite profiling to predict an individual's response to a drug. This novel approach has been termed as pharmaco-metabonomic approach82.
The ultimate objective of the microbiome research is to utilize this knowledge to predict the risk of disease development, develop newer techniques to diagnose related diseases and evolve therapeutic approaches for manipulation of the human microbiome for betterment of mankind.
References
- From meta-omics to causality: experimental models for human microbiome research. Microbiome. 2013;1:14.
- [Google Scholar]
- The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. 2012;9:577-89.
- [Google Scholar]
- Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci USA. 2002;99:15451-5.
- [Google Scholar]
- Metagenomics - a guide from sampling to data analysis. Microb Inform Exp. 2012;2:3.
- [Google Scholar]
- A metaproteomic approach to study human-microbial ecosystems at the mucosal luminal interface. PLoS One. 2011;6:e26542.
- [Google Scholar]
- Metaproteomics: studying functional gene expression in microbial ecosystems. Trends Microbiol. 2006;14:92-7.
- [Google Scholar]
- An invitation to the marriage of metagenomics and metabolomics. Cell. 2006;134:708-13.
- [Google Scholar]
- The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1:6ra14.
- [Google Scholar]
- Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev. 1995;59:143-69.
- [Google Scholar]
- Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Res. 2009;19:1141-52.
- [Google Scholar]
- Shotgun sequencing. 2013. Available from: http://en.wikipedia.org/wiki/Shotgun_sequencing
- [Google Scholar]
- Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012 2012 251364
- [Google Scholar]
- Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J Virol. 2009;83:4642-51.
- [Google Scholar]
- Characterization of the viral microbiome in patients with severe lower respiratory tract infections, using metagenomic sequencing. PLoS One. 2012;7:e30875.
- [Google Scholar]
- MetaHIT consortium. Metagenomics of the intestinal microbiota: potential applications. Gastroenterol Clin Biol. 2010;34:S23-8.
- [Google Scholar]
- Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837-48.
- [Google Scholar]
- A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59-65.
- [Google Scholar]
- Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694-97.
- [Google Scholar]
- The human microbiome and its potential importance to pediatrics. Pediatr. 2012;129:950-60.
- [Google Scholar]
- Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol. 2011;9:279-90.
- [Google Scholar]
- Towards the human intestinal microbiota phylogenetic core. Environ Microbiol. 2009;11:2574-84.
- [Google Scholar]
- Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci USA. 2011;108:4680-7.
- [Google Scholar]
- The importance of the development of the intestinal microbiota in infancy. Curr Opin Pediatr. 2009;21:794-800.
- [Google Scholar]
- Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics. 2006;118:511-21.
- [Google Scholar]
- The developing intestinal microbiome and its relationship to health and disease in the neonate. J Perinatol. 2011;31:S29-34.
- [Google Scholar]
- Detection of potentially novel bacterial components of the human skin microbiota using culture-independent molecular profiling. J Med Microbiol. 2005;54:1231-8.
- [Google Scholar]
- Molecular analysis of human forearm superficial skin bacterial biota. Proc Natl Acad Sci USA. 2007;104:2927-32.
- [Google Scholar]
- Diversity of the human skin microbiome early in life. J Invest Dermatol. 2011;131:2026-32.
- [Google Scholar]
- Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190-2.
- [Google Scholar]
- Comparative analyses of the bacterial microbiota of the human nostril and oropharynx. MBio. 2010;1:e00129-10.
- [Google Scholar]
- Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184:957-63.
- [Google Scholar]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2013;486:207-14.
- [Google Scholar]
- Analyses of the stability and core taxonomic memberships of the human microbiome. PLoS One. 2013;8:e63139.
- [Google Scholar]
- How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr. 2002;22:283-307.
- [Google Scholar]
- Bacterial regulation of intestinal immune responses. Gastroenterol Clin North Am. 2005;34:401-12.
- [Google Scholar]
- Intestinal microbiota of infants with colic: development and specific signatures. Pediatrics. 2013;131:e550-8.
- [Google Scholar]
- Neonatal inflammatory intestinal diseases: Necrotising enterocolitis and allergic colitis. Early Hum Dev. 2005;81:117-22.
- [Google Scholar]
- Early intestinal bacterial colonization and necrotizing enterocolitis in premature infants: the putative role of Clostridium. Pediatr Res. 2004;56:366-70.
- [Google Scholar]
- Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS One. 2011;6:e20647.
- [Google Scholar]
- Acute necrotizing enterocolitis of preterm piglets is characterized by dysbiosis of ileal mucosa-associated bacteria. Gut Microbes. 2011;2:234-43.
- [Google Scholar]
- Perinatal programming of asthma: the role of gut microbiota. Clin Dev Immunol 2012 2012 932072
- [Google Scholar]
- An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027-31.
- [Google Scholar]
- Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011;5:82-91.
- [Google Scholar]
- Does the human gut microbiota contribute to the etiology of autism spectrum disorders? Dig Dis Sci. 2012;57:1987-9.
- [Google Scholar]
- Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22:850-9.
- [Google Scholar]
- Characterization of skin microbiota in patients with atopic dermatitis and in normal subjects using 16S rRNA gene-based comprehensive analysis. J Med Microbiol. 2007;56:1675-83.
- [Google Scholar]
- Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS One. 2008;3:e2719.
- [Google Scholar]
- Childhood asthma after bacterial colonization of the airway in neonates. N Engl J Med. 2007;357:1487-95.
- [Google Scholar]
- Microbial profiles in saliva from children with and without caries in mixed dentition. Oral Dis. 2012;18:595-601.
- [Google Scholar]
- Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011;10:497-506.
- [Google Scholar]
- Sinus microbiome diversity depletion and Corynebacterium tuberculostearicum enrichment mediates rhinosinusitis. Sci Transl Med. 2012;4:151ra124.
- [Google Scholar]
- Virus-plus-susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135-45.
- [Google Scholar]
- Genome-virome interactions: examining the role of common viral infections in complex disease. Nat Rev Microbiol. 2011;9:254-64.
- [Google Scholar]
- Sequence analysis of the human virome in febrile and afebrile children. PLoS One. 2012;7:e27735.
- [Google Scholar]
- Comparative analysis of fecal microflora of healthy full-term Indian infants born with different methods of delivery (vaginal vs caesarean): Acinetobacter sp. prevalence in vaginally born infants. J Biosci. 2012;37:989-98.
- [Google Scholar]
- Faecal bifidobacteria in Indian neonates & the effect of asymptomatic rotavirus infection during the first month of life. Indian J Med Res. 2010;132:721-7.
- [Google Scholar]
- Toll-like receptor 9 signaling mediates the anti-inflammatory effects of probiotics in murine experimental colitis. Gastroenterology. 2004;126:520-8.
- [Google Scholar]
- A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620-5.
- [Google Scholar]
- Soluble proteins produced by probiotic bacteria regulate intestinal epithelial cell survival and growth. Gastroenterology. 2007;132:562-75.
- [Google Scholar]
- Thuricin CD, a post-translationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc Natl Acad Sci USA. 2010;107:9352-7.
- [Google Scholar]
- Antimicrobial activity of lacticin 3,147 against clinical Clostridium difficile strains. J Med Microbiol. 2007;56:940-6.
- [Google Scholar]
- Dietary modulation of the human colonic microbiota: Introducing the concept of prebiotics. J Nutr. 1995;125:1401-12.
- [Google Scholar]
- Probiotics for prevention of necrotising enterocolitis in preterm neonates with very low birthweight: A systematic review of randomised controlled trials. Lancet. 2007;369:1614-20.
- [Google Scholar]
- The utility of probiotics in the treatment of irritable bowel syndrome: A systematic review. Am J Gastroenterol. 2009;104:1033-49.
- [Google Scholar]
- Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin Infect Dis. 2011;53:994-1002.
- [Google Scholar]
- Gastrointestinal microbiome signatures of pediatric patients with irritable bowel syndrome. Gastroenterology. 2011;141:1782-91.
- [Google Scholar]
- Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature. 2006;440:1073-7.
- [Google Scholar]




