
Translate this page into:
Navigating investigator-initiated clinical trials: A call for guidelines & monitoring frameworks from an Indian context
For correspondence: Dr Aparna Mukherjee, Clinical Studies and Trials, Division of Development Research, Indian Council of Medical Research, New Delhi, 110 029, India e-mail: aparna.sinha.deb@gmail.com
-
Received: ,
Accepted: ,
Abstract
Investigator-initiated clinical trials – also known as non-regulatory or academic clinical trials, are conducted by investigators from academia or research organizations. They usually aim to address scientific questions with insufficient commercial implications and generate real-world applicable solutions, unlike trials sponsored by the pharmaceutical industry which are primarily focused on marketing approval of products that have a commercial value. For the trial results to be credible, adhering to robust methodology and the highest quality standards is paramount. Currently, investigator-initiated clinical trials in India are beyond the purview of the national regulatory authority. They are guided mainly by the National Ethical Guidelines for Biomedical and Health Research Involving Human Participants, 2017 published by Indian Council of Medical Research. They lack an accepted framework for review, conduct, monitoring, reporting of adverse events, and participant compensation. Considering this scenario, we discuss the challenges faced in an investigator initiated clinical trial and explore plausible solutions.
Keywords
Investigator-initiated clinical trial
non-commercial clinical trials
risk-based monitoring
clinical trial monitoring
academic trial

In India, clinical trials (CT) have traditionally been classified based on the sponsor type into industry-sponsored trials and investigator-initiated (non-regulatory or academic) trials. The New Drugs and Clinical Trials (NDCT) Rules, 2019 defines clinical trials in human subjects as ‘any systematic study of a new drug or investigational new drug in human subjects to generate data for discovering or verifying its (i) clinical or; (ii) pharmacological including pharmacodynamics, pharmacokinetics or; (iii) adverse effects; to determine the safety, efficacy or tolerance of such drug or investigational new drug’1. Any such trial involving a new product requires regulatory approval before the trial is conducted to make the data admissible for commercialization or marketing later1. These trials are governed by the NDCT Rules, 2019 and monitored directly by the central licensing authority - Central Drugs Standard Control Organization (CDSCO). They are hence also referred to as regulatory CTs. Industry-sponsored trials are generally of higher risk since they are often for a new product whose safety profile is largely unknown.
In contrast, investigator-initiated CTs are initiated by an individual or a group of investigators from academia or research organizations, with a focus on answering a pertinent clinical or public health query rather than with the intent of product commercialization, hence also called academic trials. NDCT rules 2019, defines academic CT as ‘a clinical trial of a drug already approved for a certain claim and initiated by any investigator, academic or research institution for a new indication or new route of administration or new dose or new dosage form, where the results of such a trial are intended to be used only for academic or research purposes, and not for seeking approval of the Central Licensing Authority or regulatory authority of any country for marketing or commercial purpose’1. There is still some ambiguity with the use of these terms because it can be argued that a regulatory CT sponsored by a pharmaceutical company also has an academic pursuit and an investigator (leading to confusion when the terms academic CT or investigator-initiated CT are used). Even academic CTs need support from the pharmaceutical industry to obtain the investigational product and/or placebo2. According to the NDCT rules 2019, the distinguishing feature between regulatory and academic CTs is whether the trial results will be used to expand the indication through regulatory filing seeking marketing approval. As a result, an acceptable phrase for consideration may be 'regulatory CT' for those that would eventually be utilized for regulatory approval of a certain indication, while others may be referred to as 'non-regulatory CT'. It should be cautioned that non-regulatory does not mean that the trial does not have an overseeing body, but it is monitored at institutional level by the ethics committee (EC) rather than by the Central licensing authority (CLA).
Investigator-initiated CTs are conventionally funded either through research grants or sometimes might not be funded at all. The principal investigator (PI) or his/her institute bears the sponsor's responsibilities in such trials. Though it is expected that the investigators will follow Good clinical practices (GCP) and the NDCT rules 2019, there lacks a defined mechanism to ensure compliance. The NDCT rules 2019 mention that any academic trial should follow the Indian Council of Medical Research (ICMR) National ethical guidelines for biomedical and health research involving human participants, 2017 (hereafter referred to as ICMR guidelines, 2017). However, these guidelines predate the NDCT Rules 2019 and do not have a definition or specific academic trial provisions. Today, any form of national guideline for conduct, monitoring, and quality assurance of investigator-initiated CTs is largely lacking. The studies conducted by the pharmaceutical industry for regulatory approval are intensely monitored, and steps are taken to ensure the robust quality of the studies. Since investigator-initiated trials are often of lower risk, they should not be burdened with overwhelming paperwork but must have systems for ensuring the safety of the participants and the sanctity of the study. A middle path can be adapted from existing systems for regulatory CT.
Investigator-initiated CTs have been instrumental in answering questions that may not be of interest to the pharmaceutical industry but are critical for the advancement of clinical management of patients and the improvement of public health3. The relevance of investigator-initiated CTs was increasingly felt when various approved medicines were repurposed for COVID-19. The current paper highlights the challenges faced while conducting an investigator-initiated CT and suggests potential solutions that could be of help in standardizing the conduct of such trials, making them more scientifically robust.
Current landscape of investigator-initiated clinical trials in India
In India, between January 1, 2006 to December 31, 2017, 61 per cent of 3138 trials registered at the Clinical Trial Registry of India were industry-funded4. Investigator-initiated trials are important for the advancement of medical science as they are usually driven by scientific curiosity, academic enquiry, and public health imperatives. Even though investigator-initiated trials utilize medicines approved for use, they are not entirely devoid of risks. Hence, high standards of review and oversight are essential for data integrity and participant safety. Some of the concerns and challenges the authors faced while conducting and reviewing investigator-initiated CTs are shared here.
Need for robust scientific review: A system of thorough scientific review of an investigator-initiated CT protocol is in place in many institutes that carry out such trials. However, an unbiased, robust review which will improve the protocol and identify the risks involved may not be uniformly available to all investigators or institutes undertaking such CTs. A scoping review evaluating the quality and effectiveness of ethics review identified that most studies evaluated the administrative aspects of review committees (such as membership and constitution, timelines for approval, cost, workload, etc.). At the same time, very few studies looked at functional outcomes (such as patterns of agreement in decision, improved patient outcomes, improved knowledge, etc5,6. Further research is also needed to better understand the robustness of review processes of the EC.
No clear mechanism for oversight: In India, investigator-initiated CTs do not need regulatory clearance from the central licensing authority unless deemed necessary by the EC, based on assessing the risks to the trial participants. After the CDSCO is informed about an academic trial, if no reply is received within 30 days, it can be assumed that permission is not required. Like any other research involving human participants, review and approval from the EC is mandatory. However, ECs are sometimes averse to the associated risks of CTs. Also, many of the ECs lack the bandwidth in terms of human resources and relevant expertise or experience to review and monitor a CT. It may be further noted that many ECs have limited support from the institutions to undertake proper physical monitoring5-8.
Poor documentation practices: As per NDCT rules 2019 and GCP Guidelines, regulatory CT in India requires meticulous documentation to ensure compliance with regulatory requirements, protect participant safety, and maintain data integrity. The amount of paperwork may dissuade many investigators, and essential documents are sometimes not maintained due to unawareness or inertia from lack of oversight. Space constraints also add to the problem of record-keeping and archival9. With no guiding rules or a third-party overview, it is presumably upon the PI, the host institute and the relevant EC to be responsible for the practices followed in the trial.
No clear mechanism for causality assessment in case of serious adverse events (SAEs) and compensation to trial-related injury: The frameworks and timelines for reporting SAEs are not adequately described for investigator-initiated trials, and neither are rules concerning compensation for injury or death. However, ECs are encouraged to have Standard Operating Procedures (SOPs) related to these aspects. The reporting and calculation are provided in the NDCT rules and are enforceable for regulatory trials. However, investigator-initiated trials are not regulated by these guidelines. The ICMR guidelines 2017 state that any SAE must be reported within 24 h and also that the compensation for the participants in case of a trial is the responsibility of the host institute. However, it is unclear who will assess the causality of SAE, and how the compensation will be calculated. Additionally, most institutes do not have the kind of corpus required to compensate CT subjects in case of a SAE. Some ECs are known to mandate clinical trial insurance1,10.
No clear mechanism for monitoring: The responsibility of monitoring an investigator-initiated CT rests with the EC, which has approved the study as per the ICMR guidelines 201710. However, the guidelines do not describe how this monitoring will be operationalized. It is also unclear how multi-centre investigator-initiated trials will be steered. This depends on whether the local EC is equipped with the requisite capacity and experience to provide risk-proportionate monitoring to the ongoing trials, even more so in multicenter trials. Additionally, an EC may be overburdened with monitoring several trials approved within the same period. Currently, the monitoring provided by most ECs for investigator-initiated CTs is limited to assessing the relatedness of the SAEs to the trial procedures and reviewing a final study report; which is arguably not enough to make the study monitoring fair and adequate.
Poor quality control: The fundamental premise of any academic research is to answer a pertinent problem and find solutions that are translatable to practice or policy. For the findings of investigator-initiated trials to be credible and usable, there is a need for robust quality assurance practices throughout the trial lifecycle. To ensure that the same quality standards as in a regulatory trial are maintained, quality assurance mechanisms are required to be in place, which are often non-existent.
Suggestions for improving investigator-initiated or academic clinical trials in India
Given the above-mentioned challenges, the authors propose the following suggestions that could help investigators of investigator-initiated CTs to make their study more rigorous, trustworthy, and impactful.
Invest in research review capacity: Institutions that plan to undertake investigator-initiated CTs must set up relevant review committees for an unbiased and thorough scientific review of all study protocols. This will help identify any concerns and improve the participant's safety, scientific integrity, and usefulness of the study. Accreditation and registration of ECs are expected to improve their quality and effectiveness11,12. The accreditation programmes are an attempt to standardize the quality of clinical research in India, and ensures public trust and confidence in the ethics committees. Clear standards for the composition of the ethics committee, protection of the subject's rights, safety and well-being, review process and monitoring of clinical trials make a committee more committed to quality and accountability. Accreditation in India can be done at a minimal fee, which must be updated annually13.
Define oversight framework: The ethics review process is well documented in the ICMR guidelines 2017, which can be used to refer to the review of an investigator-initiated trial proposal. Though not mandatory for investigator-initiated trials, it is preferable to have the EC within 50 km of the institute, for ease of monitoring. It is also advisable to have the EC registered with CDSCO and the Department of Health Research, and its composition may be aligned with the NDCT rules 2019. Institutes conducting investigator-initiated trials must invest more resources to their ECs, ensuring that all members are adequately trained and remain abreast with the latest developments. ECs may also require the investigators to submit quarterly reports to review ongoing trials instead of annual reports.
Improve documentation practices: Maintaining a Trial master file (TMF) with the essential documents is a good practice for all investigators to inculcate. NDCT 2019 provides clear guidelines on the components of TMF, which include all requisite approval, trial protocol and accessory documents, sponsor details, contracts, regulatory documents, investigators brochures, etc. Even though there are plenty of references regarding the essential documents for a regulatory trial, there is no clarity on what documents must be maintained for an investigator-initiated CT. Some of the adaptations that can be applied are combining documents like screening and recruitment logs; study drug having marketing authorization and obtained via routine medicines supply chain like pharmacies or stores may not need detailed accountability documents14.
Develop a framework for reporting and evaluating SAE causality & compensation for trial-related injury: Considering several trials would be concomitantly approved by an EC, it might not always be possible to have a fair review of the relatedness of SAE to the trial procedures by the same committee. This may be due to various concerns, including the unavailability of a subject expert in the committee, or lack of time and human resources. Hence, we propose that an independent causality assessment committee examine the SAEs for a fair, independent, and timely assessment. This committee would also decide upon the compensation to be provided to the participant in case the SAE is related to the trial. The members of this committee can include a pharmacologist, one or two subject experts, a biostatistician, and a member of the EC. This causality assessment committee can also double as the data and safety monitoring board, as described below. One such committee could be appointed for a designated tenure and could cater to the needs of multiple trials, under the same domain. The members of the committee should declare their conflict of interests. A similar framework has been followed in the ongoing RECOVERY International trial at the India sites15.
Figure 1 gives a flow of SAE reporting that can be followed in case of an investigator-initiated trial. The timelines of reporting are similar to those observed by the CDSCO for a regulatory trial. Additionally, based on the risk level of the trial, it would be prudent to either have a corpus fund for compensation or procure a clinical trial liability insurance, which could be budgeted in the study budget while submitting it to the funding agency.
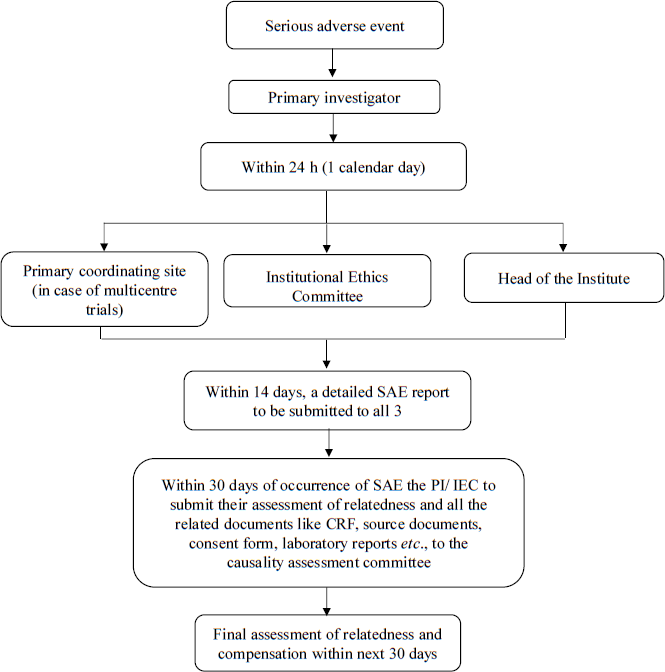
- Proposed timelines of reporting a Serious Adverse Event (SAE). PI, principal investigator; IEC, Institutional Ethics Committee; CRF, case record form.
Develop a framework for monitoring: The International Conference on Harmonization of Good Clinical Practice (ICH GCP) defines trial monitoring as ‘the act of overseeing the progress of a clinical trial and of ensuring that it is conducted, recorded and reported in accordance with the protocol, Standard Operating Procedures (SOPs), Good Clinical Practice (GCP), and the applicable regulatory requirement(s)16. It does not define the exact monitoring mechanism, but guides that the extent and nature of monitoring should be individualized based on the objective, purpose, design, complexity, blinding, size, and endpoints of the trial; and the monitors should be appropriately trained, should have requisite qualifications, should be familiar with the investigational product and finally well versed with the study protocol16.
The concept of risk-proportionate monitoring and risk-based quality management of CT is being increasingly accepted worldwide17. The purpose of risk assessment is to maintain the quality of the study procedures and the data integrity, while also mitigating any potential risks to the study participants. Risks in a CT can be attributed to both the intervention and the trial itself. Interventions can be of low risk when they are minimally different from acceptable clinical practice or high risk when they are novel entities. Trials including vulnerable groups, patients with severe/novel diseases, complex design, larger sample size, complicated and frequent procedures, etc., may be labelled as high risk. In case of a multi-centre trial, a site experiencing high screening failure and slow recruitment might need special monitoring to identify bottlenecks18. Consequently, the ‘one size fits all’ approach may not be appropriate for all trial monitoring and the content of the trial monitoring plan may vary from trial to trial, and is also dynamic where monitoring components could change over the course of the trial.
Investigator-initiated trials, by nature, are supposed to be of lower risk than regulatory ones, even though this need not always be the case. Hence, the risk-proportionate monitoring or risk-based monitoring will work very efficiently in this scenario. This will enable the investigators to develop a plan that is suitable and commensurate with the risks involved in their trials while at the same time preventing the teams from being overburdened with monitoring tasks. A prospective, stratified, cluster-randomized study comparing extensive on-site monitoring with risk-adapted monitoring included 213 sites from 11 investigator-initiated trials. The ADAMON study found that risk-adapted monitoring utilized half the resources, and was non-inferior to extensive on-site monitoring, suggesting that monitoring in clinical trials can be effectively managed without the extensive resource commitment traditionally associated with comprehensive monitoring strategies19.
Figure 2 summarizes how risk-based monitoring could be undertaken in an investigator-initiated trial. There are many risk assessment tools available20 and four basic steps in risk-based monitoring (Fig. 2). The principles of effective and efficient risk-based monitoring are described below using these four steps, along with a proposal of who could monitor investigator-initiated trials:
-
(i)
Identifying the critical data points and processes: At the onset of a study, the PI and the study team need to identify the risks involved. An initial risk assessment will involve multiple stakeholders who will identify Critical to Quality (CtQ) risks across the life cycle of a trial. The multidisciplinary team of stakeholders need to decide upon as to what are the critical processes, such as the verification of informed consent, strict adherence to the inclusion/exclusion criteria, maintaining randomization and blinding, and identification of critical data parameters such as adverse events, outcome assessment and SAEs. The likelihood of the identified risk occurring and its impact on patient safety and data quality will determine the level of risk.
-
(ii)
Assessment of risk: Monitoring of high-risk trials could be conducted more frequently and on-site in case of multi-centre studies21. Risks must be assessed based on the following factors:
-
(a)
Complexity of study designs: Adaptive studies, stratified designs, studies requiring dose titrations could require more intensive monitoring.
-
(b)
Study end points: Objective endpoints such as death and hospitalization are more straightforward, while endpoints requiring interpretation, such as chest X-ray reading or symptom resolution may need an independent blinded assessor or adjudication committee.
-
(c)
Vulnerable population: A trial involving vulnerable populations or serious illnesses should be considered for more scrutiny to ensure that appropriate patient safety measures are followed.
-
(d)
Experience of the PI/Institute/study staff: Investigators lacking experience could be more closely monitored on-site, especially if the trial involves surgical procedures, medical devices, severe patients, vulnerable populations, etc. This ensures that such trials receive due diligence and mentoring support by helping investigators transparently address operational and technical difficulties.
-
(e)
Stage of the trial: It might be more useful to monitor study initiation activities and taper it over a period.
Many other trial-specific factors could be used in risk assessment.
-
-
(iii)
Development of risk-based monitoring plan: Mitigation strategies are to be designed to assess the risk level of the identified critical points. A monitoring plan should be in place before the trial initiation and include at least the following points:
-
(a)
Brief description of the study: The description should include objectives, process, and the identified critical processes and data points.
-
(b)
Monitoring approach: Specific risks must be addressed along with the monitoring method, timing, frequency, and extent. It should also include the required tools, logs and documentation. Additionally, it should clarify how deviations or failures will be dealt with.
-
(c)
Communication strategy: The reporting mechanisms of the results, frequency, formats, contents, etc. need to be specified.
-
(d)
Management of noncompliance: Strategies for addressing noncompliance with the study protocol should be elaborated.
-
(e)
Quality assurance mechanisms: All personnel involved in monitoring activities should be appropriately trained on the principles of clinical investigations involved, trial design, protocol, data collection techniques, monitoring methods, etc.
-
(f)
Monitoring multi-centre trial: Risk-based monitoring ideally needs to be centralized with all data flowing in a central dashboard that will facilitate easy access and quick corrective measures when required22. For trials where such central data monitoring is not possible, a source document verification of a certain proportion of critical data points may be considered.
-
-
(iv)
Ongoing risk assessment: Risk assessment should be continuous throughout the trial. New evidence obtained from published literature or ongoing studies can help modify the risk assessment and mitigation plan. Certain pre-defined tolerance limits for the risk parameters must be listed before the trial's initiation. These limits will trigger the need for more aggressive corrective actions. Previous studies identified certain aspects or tasks that could be critical for monitoring9,23. The Table presented here highlights a few examples of the process of risk-based monitoring.
-
(v)
Who will conduct the monitoring: The ICH-GCP guidelines mention that ‘The IRB/IEC should conduct continuing review of each ongoing trial at intervals appropriate to the degree of risk to human subjects, but at least once per year.’ The same is also suggested by the ICMR guidelines 2017, which says, ‘ECs are entrusted with the initial review of research proposals prior to their initiation, and also have a continuing responsibility to regularly monitor the approved research to ensure ethical compliance during the conduct of research’. The guidelines by ICMR do not specifically discuss investigator-initiated CT and the focus of monitoring is more on ethical compliance, rather than the overall conduct of study. Hence, there is currently no clarity regarding who will monitor an investigator-initiated CT.
| Identified critical points and processes | Potential impact on study | Likelihood of occurring | Mitigation strategy |
|---|---|---|---|
| Improperly obtained informed consent | Will jeopardize the rights of participants | High |
Adequate training of personnel; may involve role-playing for better results Standardized operating procedures The process being observed in a few instances Refresher training for research staff |
| Deviation from inclusion/exclusion criteria | Safety of participants as well as validity of results adversely affected | Moderate |
Appropriate population selected for screening Checklists applied |
| Improper storage of study product | Safety of participants compromised/results may not be accurate | Moderate |
Ensure proper area/equipment for storage are available before initiation of study Maintenance of temperature logs |
CT, clinical trials
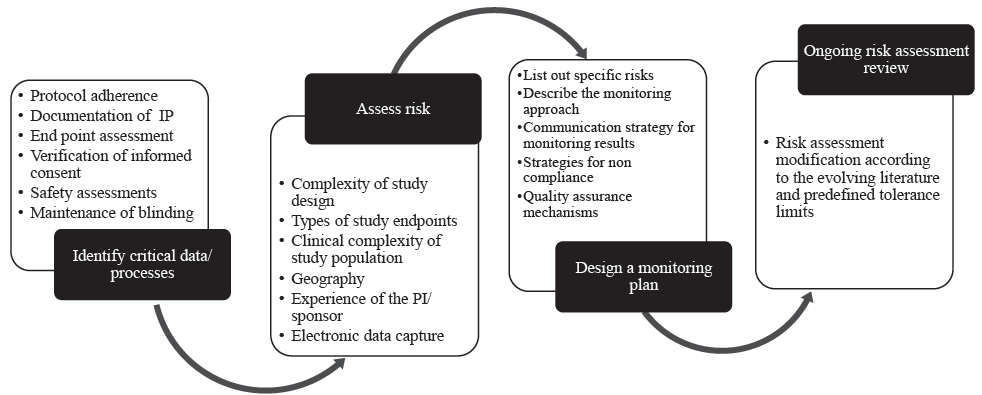
- Process of how risk-based monitoring of a CT could be undertaken in an investigator-initiated trial. IP, investigational product.
One of the following strategies could be considered based on the resources available to the EC and the researcher:
-
(i)
An independent trial specific committee: Such a committee may be constituted with external and/ or internal members from the institute, with one person from the approving EC, if needed. All committee members should have pre-defined documented responsibilities. The constitution and formation of this committee would be the responsibility of the PI, and it could be supervised by the EC of the institute. Such peer mentoring will benefit all the investigators in a region in the long run24.
-
(ii)
A subcommittee of the EC: A few members from the EC and other members co-opted as needed may constitute this committee. This would divide the burden of monitoring many CTs among the members of the EC as well as expand the expertise pool of the monitoring committee. The formation and functioning of this subcommittee would be guided by the core members of the EC. A similar approach was followed in the past by one of the institutes of India where 14 trials were monitored by a five-member subcommittee created by the EC and the monitoring focused on documentation and record practices, participant’s rights and compliance with the protocol, and storage and access of the trial supplies. Several discrepancies were identified by this subcommittee, which further emphasized the importance of this activity8.
-
(iii)
Institutional trial monitoring unit: The institutes that conduct investigator-initiated CTs could form a structured clinical trial/research unit with a pool of clinical trial monitors, which would have personnel of relevant expertise, specifically hired for the purpose of monitoring CTs. These monitors will meet the investigators at a pre-defined frequency and would be responsible for continuous review, monitoring consent process, ensuring protocol adherence and data integrity24. They would also ensure that the documentation and approvals are up to date and the investigational product is managed and stored properly.
-
(iv)
Monitoring committee constituted by the funder: The responsibility of monitoring activity could also be taken up by the funding agency of the concerned trial, especially in large, multi-center trials. The monitoring by the funding agency may preferably be in collaboration with the local EC, to avoid redundancy. However, this may be possible only for CTs with high risk or national priority.
Improving quality and data integrity
Traditionally, all regulatory trials have a Data and Safety Monitoring Board (DSMB) or an Independent Data Monitoring Committee (IDMC). The DSMB, appointed by the sponsor, is an independent committee comprising independent experts in relevant fields, pharmacologist and biostatistician and provides unbiased oversight and monitoring throughout the trial25. The primary functions of the DSMB in CT include safety monitoring (analyzing data pertaining to AEs/SAEs); interim analysis of the study, if required; assessment of risk and benefit; addressal of significant protocol violations; recommending any protocol changes based on safety data; and ensuring data integrity. Constituting a DSMB for investigator-initiated trials is a good check mechanism that can be adopted by investigators and institutions to improve the quality of their studies. It should be considered that the DSMB is independent and does not include people from the institute. The guidelines for funding of DSMB in India are limited, though the WHO and NIH guidelines mention that DSMB members should be paid an honorarium and travel expenses, which could be budgeted in the study26,27.
Conclusion
Investigator-initiated or academic CTs are important tools for the betterment of medical knowledge as well as clinical practice. A more structured approach with some checks on the conduct and appropriate monitoring, will enhance the credibility and robustness of the data generated. This approach will also build confidence in the stakeholders about safeguarding the interests and rights of all the participants. At the same time, individual researchers should not be overburdened with the complexity of record-keeping, and a rational, risk-appropriate approach is recommended.
Financial support & sponsorship
None.
Conflicts of Interest
None.
Use of Artificial Intelligence (AI)-Assisted Technology for manuscript preparation
The authors confirm that there was no use of AI-assisted technology for assisting in the writing of the manuscript and no images were manipulated using AI.
References
- New Drugs & Clinical Trial Rules. Available from: https://cdsco.gov.in/opencms/export/sites/CDSCO_WEB/Pdf-documents/NewDrugs_CTRules_2019.pdf, accessed on April 2, 2024.
- Investigator initiated trials (IITs) Perspect Clin Res. 2012;3:119-21.
- [CrossRef] [PubMed] [Google Scholar]
- Impact of investigator initiated trials and industry sponsored trials on medical practice (IMPACT): Rationale and study design. BMC Med Res Methodol. 2020;20:246.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- New clinical trial rules: Academic trials and tribulations. Perspect Clin Res. 2019;10:103-5.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Ethics review of multi-centre clinical trials in Canada. Health Law Rev. 2005;13:51-7.
- [PubMed] [Google Scholar]
- A systematic review of the empirical literature evaluating IRBs: What we know and what we still need to learn. J Empir Res Hum Res Ethics. 2011;6:3-19.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Ethics committees in India: Facing the challenges! Perspect Clin Res. 2012;3:50-6.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- On-site monitoring of clinical trials by an Ethics Committee in India: A road less travelled. Res Ethics. 2021;17:45-54.
- [CrossRef] [Google Scholar]
- Compliance of Mumbai-based clinical trial sites with the Quality Council of India guidelines and evaluation of the challenges faced by the investigators. Perspect Clin Res. 2021;12:133-9.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- National ethical guidelines for biomedical and health research involving human participants. Available from: https://ethics.ncdirindia.org/asset/pdf/ICMR_National_Ethical_Guidelines.pdf, accessed on November 28, 2023.
- Literature Review: Quality standards and accreditation schemes for human research ethics committees. Available from: https://www.safetyandquality.gov.au/publications-and-resources/resource-library/literature-review-quality-standards-and-accreditation-schemes-human-research-ethics-committees-0, accessed on April 14, 2024.
- Ethics committees: Challenge of evidence-based accreditation. Perspect Clin Res. 2017;8:105-6.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Ethics Committee Accreditation Programme - NABH. Available from: https://nabh.co/clinical-trial/, accessed on July11, 2024.
- Risk proportionate approaches in clinical trials. Available from: https://health.ec.europa.eu/system/files/2017-08/2017_04_25_risk_proportionate_approaches_in_ct_0.pdf, accessed on April 2, 2024.
- Asia and Africa. Available from: https://www.recoverytrial.net/international/international-sites, accessed on December 7, 2023.
- Integrated addendum to ich e6(r1): guideline for good clinical practice e6(r2). Available from: https://database.ich.org/sites/default/files/E6_R2_Addendum.pdf, accessed on July 11, 2024.
- Risk-based monitoring in clinical trials: 2021 update. Ther Innov Regul Sci. 2023;57:529-37.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Risk-proportionate clinical trial monitoring: an example approach from a non-commercial trials unit. Trials. 2014;15:1-10.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Risk-adapted monitoring is not inferior to extensive on-site monitoring: Results of the ADAMON cluster-randomized study. Clin Trials. 2017;14:584-96.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The risk based monitoring toolbox. Available from: https://ecrin.org/risk-based-monitoring-toolbox, accessed on April 2, 2024.
- Generating evidence on a risk-based monitoring approach in the academic setting – lessons learned. BMC Med Res Methodol. 2017;17:26.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Risk-based monitoring in clinical trials: Past, present, and future. Ther Innov Regul Sci. 2021;55:899-906.
- [CrossRef] [PubMed] [Google Scholar]
- Common deviations related to data-handling, etc. in investigator-initiated trials. Available from: https://laegemiddelstyrelsen.dk/en/licensing/clinical-trials/gcp-inspection/common-deviations-related-to-data-handling,-etc-in-investigator-initiated-trials/, accessed on April 5, 2024.
- Assuring data quality in investigator-initiated trials in Dutch hospitals: Balancing between mentoring and monitoring. Account Res. 2022;29:483-511.
- [CrossRef] [PubMed] [Google Scholar]
- Available from: https://tdr.who.int/publications/m/item/2005-02-01-operational-guidelines-for-the-establishment-and-functioning-of-data-and-safety-monitoring-boards, accessed on October 4, 2024.
- Guidelines for establishing and operating a data and safety monitoring board. Available from: https://www.hhs.gov/guidance/document/guidelines-establishing-and-operating-data-and-safety-monitoring-board, accessed on April 14, 2024.
- Data and Safety Monitoring Board (DSMB) Guidelines. Available from: https://www.nidcr.nih.gov/research/human-subjects-research/toolkit-and-education-materials/interventional-studies/data- and-safety-monitoring-board-guidelines, accessed on July 11, 2024.




