
Translate this page into:
Identification of steroidal cardenolides from Calotropis procera as novel HIV-1 PR inhibitors: A molecular docking & molecular dynamics simulation study
For correspondence: Dr Estari Mamidala, Department of Zoology, Kakatiya University, Warangal 506 009, Telangana, India e-mail: drestari@kakatiya.ac.in
-
Received: ,
Abstract
Background & objectives
Despite advancements in antiretroviral therapy, drug-resistant strains of HIV (human immunodeficiency virus) remain a global health concern. Natural compounds from medicinal plants offer a promising avenue for developing new HIV-1 PR (protease) inhibitors. This study aimed to explore the potential of compounds derived from Calotropis procera, a medicinal plant, as inhibitors of HIV-1 PR.
Methods
This in silico study utilized natural compound information and the crystal structure of HIV-1 PR. Molecular docking of 17 steroidal cardenolides from Calotropis procera against HIV-1 PR was performed using AutoDock 4.2 to identify compounds with higher antiviral potential. A dynamic simulation study was performed to provide insights into the stability, binding dynamics, and potential efficacy of the top potential antiviral compound as an HIV-1 therapeutic.
Results
We found that all tested cardenolides had higher binding affinities than Amprenavir, indicating their potential as potent HIV-1 PR inhibitors. Voruscharin and uscharidin displayed the strongest interactions, forming hydrogen bonds and hydrophobic interactions with HIV-1 PR. Voruscharin showed improved stability with lower RMSD (Root Mean Square Deviation) values and reduced fluctuations in binding site residues but increased flexibility in certain regions. The radius of gyration analysis confirmed a stable binding pose between HIV-1 PR and Voruscharin.
Interpretation & conclusions
These findings suggest that Calotropis procera could potentially be a source of compounds for developing novel HIV-1 PR inhibitors, contributing to the efforts to combat HIV. Further studies and clinical trials are needed to evaluate the safety and efficacy of these compounds as potential drug candidates for the treatment of HIV-1 infection.
Keywords
Calotropis procera
dynamic simulation
in silico
molecular docking
steroidal cardenolides
uscharidin
voruscharin

Human immunodeficiency virus (HIV) is an RNA virus that attacks the human immune system, causing acquired immunodeficiency syndrome (AIDS). Despite significant advancements in antiretroviral therapy, HIV remains a major public health concern, with over 38 million people living with the virus globally1. The current treatment of HIV involves a combination of drugs that inhibit different stages of the virus life cycle, including protease inhibitors, reverse transcriptase inhibitors, and integrase inhibitors. However, the emergence of drug-resistant strains of HIV and the side effects of current drugs highlight the need for the development of new and effective therapeutics2. The identification of new and effective HIV-1 protease inhibitors is crucial in combating drug-resistant strains of HIV and improving the efficacy of current antiretroviral therapy3.
Natural products obtained from medicinal plants have been a source of drug discovery for several decades. C. procera is a traditional medicinal plant widely distributed in different parts of the world and is commonly known as Apple of Sodom or Akund. It is a perennial shrub belonging to the Apocynaceae family. The plant is characterised by its large, fleshy leaves, bell-shaped flowers ranging from white to purple, and distinctive fruit pods containing numerous seeds with silky hairs that aid in wind dispersal. This plant has been reported to possess various pharmacological properties, including antiviral activity against HIV4. The active compounds in C. procera have been reported to exhibit antiviral activity, but their mechanism of action is not yet fully understood. The steroidal cardenolides of C. procera were selected for study due to their potent biological activities and the lack of previous research on their antiviral activity, particularly against HIV-1 PR. Recent studies have highlighted the potential of natural compounds as a source of new and effective HIV-1 protease inhibitors4. For instance, a molecular docking study by Alqahtani et al5 identified several natural compounds with potential inhibitory activity against HIV-1 protease. Similarly, a study by Salem et al6 reported the antiviral activity of a natural compound, 3-O-methylfunicone, against HIV-16. However, there is still a lack of knowledge about the antiviral activity of natural compounds from C. procera against HIV-1 protease. Thus, the present study aimed to address this research gap by identifying potential C. procera compounds to inhibit HIV-1 protease through molecular docking simulation.
Molecular docking is a computational technique widely used in drug discovery to predict the binding strength and orientation of small molecules with target proteins7. HIV-1 protease is a key enzyme required for the replication of the virus, and its inhibition is an effective strategy for treating HIV8. The protease enzyme plays a critical role in the final stages of the virus’s life cycle by cleaving viral polyproteins into functional proteins required for the maturation and replication of the virus. Thus, inhibiting the activity of HIV-1 protease can prevent the maturation and release of new virus particles, thereby reducing the viral load in the body and slowing down the progression of HIV infection. The present study utilized molecular docking and dynamic simulation to screen the 17 steroidal cardenolides of C. procera against HIV-1 protease.
Material & Methods
This study was undertaken at the department of Zoology, Kakatiya University, Telangana, India from February 2022 to January 2023.
Preparation of ligands
The cardenolides from C. procera were selected for this study to check their binding affinity with HIV-1 protease, specifically focusing on 17 steroidal cardenolides due to the lack of previous efficacy studies against HIV-1 PR and the availability of their chemical structures in the PubChem and IMPATT database. To prepare the ligands for molecular docking simulations, the 3D structures of Uscharidin, Calactin, Voruscharin, Calotropogenin, Uzarigenin, Coroglaucigenin, Syriogenin, Ascleposide, Calotropin, 7β,14 Dihydroxy-5α-Card-20(22)-Enolide, Uscharin, Beta-Anhydroepidigitoxigenin, Proceroside, Corotoxigenin, Frugoside, Afroside, Procesterol and Standard (Amprenavir-APV) were retrieved from PubChem, ( https://pubchem.ncbi.nlm.nih.gov/) and prepared for docking using Autodock software.
The ligands were downloaded in SDF (Structure Data File) format from PubChem and converted to PDB (Protein Data Bank) format using Open Babel software ( https://openbabel.org/). The ligands were further prepared for docking using Autodock Tools (ADT) software. The ligands in PDB format were converted to PDBQT (Protein Data Bank, Partial Charge-Q, and Atom Type-T) format using the AutoDockTools (ADT) software. The PDBQT format included additional information required by AutoDock for molecular docking simulations, such as atomic charges, atom types, rotatable bonds, and other parameters. The ligands were optimized using molecular dynamics simulations or quantum mechanical calculations to refine their conformations and minimise their energies.
Preparation of HIV-1 protease
The X-ray crystal structure of HIV-1 PR (PDB ID: 3NU3) was retrieved from the RCSB (Research Collaboratory for Structural Bioinformatics) Protein Data Bank9 (Fig. 1). The HIV-1 PR was loaded into ADT software for further preparation. The ligands and water molecules from HIV-1 PR were removed, as required before the molecular docking simulation. This was done using ADT, which allowed the user to remove the unwanted molecules from the protein structure. Hydrogen atoms were added to enhance the accuracy of the protein structure. Charges were assigned using ADT, incorporating Kollman charges and Gasteiger charges10. Kollman charges consider the influence of surrounding atoms on charge distribution, while Gasteiger charges are calculated based on atomic electronegativity and neighbouring atoms. The grid-based approach was employed, and grid box positions were determined based on the original protein inhibitors (APV for HIV-1 PR).
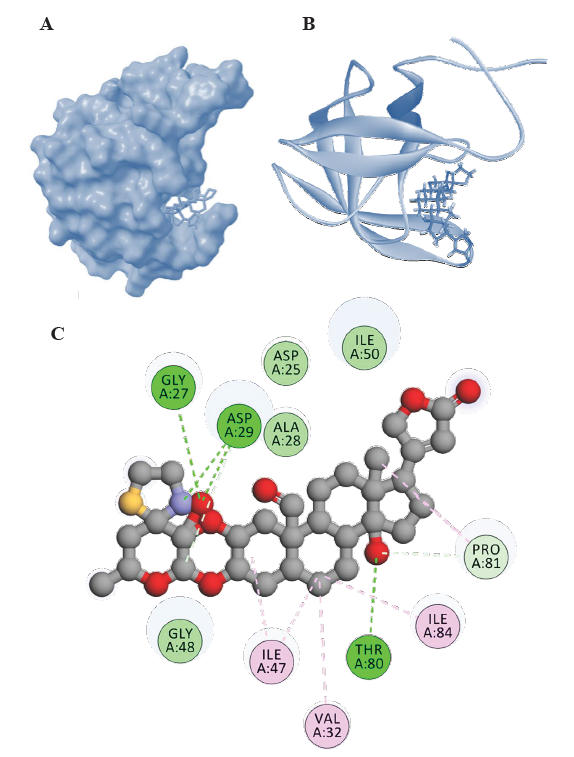
- Representations of HIV-1 PR-Voruscharin complexes from molecular docking visualized using Biovia Discovery Studio visualizer (
https://discover.3ds.com/discovery-studio-visualizer-download). (A) Surface representation of binding confirmation of HIV-1 PR with Voruscharin, (B) 3D diagram of binding conformation of HIV-1 PR with Voruscharin and (C) 2D diagram of binding conformation of HIV-1 PR with voruscharin along with the Hbonds formed in the complex and hydrophobic interactions between HIV-1 PR and voruscharin in the docked complex. Asp, aspartic acid; Gly, glycine; Thr, threonine; Ala, alanine; Asp, aspartic acid, Gly, glycine; ile, Isoleucine; Phe, phenylalanine; Val, valine; Arg, arginine; Ile; isoleucine, Met, methionine; Pro, proline.
Molecular docking simulation
To predict the active compound(s) from C. procera that block the viral enzymes activity, the computational simulation was performed using AutoDock 4.0 molecular docking software. The docking simulations were performed using the Lamarckian genetic algorithm with default parameters. The docking results provided 10 different conformations, but the conformation with the lowest binding energy was selected for further studying the protein-ligand interactions using the Discovery studio visualizer. The results were compared to APV, the original ligands of HIV-1 PR protein complex, respectively.
Molecular dynamic simulations
Molecular dynamics simulation is an exceptional technique for forecasting the characteristics and movement of novel particles11. To validate the stability outcomes of the docked complex in this study, molecular dynamics simulation was utilized to analyze the dynamic interactions between HIV-1 protease and the ligands. To carry out the molecular dynamics simulations and MM-PBSA calculations, we followed a methodology similar to a previous study12.
The HIV-1 protease and candidate molecules in the protein-ligand complex structure were prepared for molecular dynamics (MD) simulation using VMD. The system was equilibrated using NVT (Number of Particles, Volume, and Temperature) and NPT (Number of Particles, Pressure, and Temperature) ensemble for 100 ps, with trajectories generated every 2 fs and saved every 2ps. The GROMACS-2019 version ( https://manual.gromacs.org/2019/download.html) was utilized to conduct 10ns simulations with the OPLS force field. The TIP3P water model was chosen to solvate the complexes, followed by the addition of ions to neutralize the overall charge of the system. Periodic boundary conditions were employed, and energy minimization was carried out with a tolerance of 1000kJ/mol/nm. Finally, the results of the protein-ligand complex simulation were analysed. The molecular dynamic simulations were analysed by assessing the Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), and Radius of Gyration (Rg) values over time. These parameters were used to study the conformational changes, flexibility, and compactness of the system throughout the simulations.
Results
Molecular docking analysis
The Table provides the binding energies of 17 steroidal cardenolides and a standard drug (APV). Binding energy is a measure of the strength of the interaction between a ligand and its target receptor. A lower binding energy indicates a stronger binding interaction and stronger binding affinity. Based on the results in the Table, it can be observed that all the steroidal cardenolides have higher binding affinity than the standard drug (APV). The binding energies of steroidal cardenolides being significantly lower than that of APV (-12.38 to -9.04 Kcal/mol) suggested that the steroidal cardenolides had a higher affinity for their respective target receptor i.e., HIV-1 PR than APV (-6.77 Kcal/mol).
| Name of the compound | Binding energy (Kcal/Mol) | No. of hydrogen bonds | Hydrogen bond interactions | Hydrophobic interactions |
|---|---|---|---|---|
| Voruscharin | −12.38 | 2 | Asp:29, Gly:27, Thr:80 | Ala:28, Asp:25, 30, Gly:48, 49, Ile:50, Phe:53, Val:82 |
| Uscharidin | −11.32 | 2 | Arg:8, Gly:48 | Gly:27, 49, Val:32, 82 Ile:47, 50, 84, Phe:53, Asp:25, 30, Ala:28, Asp:29, Met:46, Thr:80, Pro:81 |
| Beta-Anhydroepidigitoxigenin | −10.94 | 2 | Gly:51, Asp:30 | Gly:48, 49, 52, Ile:50, Asp:29, Ala:28, Phe:53, Thr:80, Ile:47, 54, 84, Val:32, Pro:81, Asp:30 |
| Afroside | −10.88 | 3 | Arg:8, Asp:29, Gly:48 | Ile:47, 50, 84 Val:32, 82, Gly:49, Pro:81, Thr:80, Ala:28, Phe:53 |
| Calactin | −10.78 | 4 | Arg:8, Thr:80, Asp:30, Gly:48 | Leu:23, 76, Ala:28, Asp:29, Val:32, Pro:81, Val:82, Ile:84, 47, 50, 54 |
| Uscharin | −10.59 | 3 | Ile:50, Lys:49, Gly:48 | Arg:8, Gly:49, Asp29, Asp:30, Ile:47, 84, Ile:54, Val:32, 82, Pro:81, Ala:28 |
| Uzarigenin | −10.54 | 2 | Asp:29, 30 | Thr:80, Phe:53, Pro:81, Gly:48, 49, 51, 52, Ile:47, 50, 54, 84, Val:32 |
| Ascleposide | −10.37 | 2 | Arg:8, Asp:30 | Leu:23, Ile:50, 54, Gly:48, 49, 52, Ala:28, Leu:76, 54, Val:32, Thr:80, Lys:45, Asp:29, Ile:47, 84, Pro:81, Val:82 |
| Calotropin | −10.33 | 3 | Asp:29, Gly:48, Arg:8 | Val:82, Pro:81, Ile:47, 50, 84, Gly:49, Ala:28, Asp:25, Gly:27, Thr:80, Val:32, Phe:53 |
| Syriogenin | −9.87 | 3 | Asp:29, 30,Ile:50 | Gly:48, 51, 27, Asp:25, Ala:28, Gly:49, Pro:81, Leu:76, Thr:80, Pro:79, Phe:53, Gly:52, Ile:47, 54, 84, Val:32 |
| Frugoside | −9.76 | 3 | Arg:8, Asp:29, Gly:48 | Leu:23, Gly:27, 49, Ile:47, 50, 54, 84, Thr:80, Val:32, Asp:30, Leu:76, Val:82, Pro:81, Ala:28 |
| Proceroside | −9.75 | 3 | Arg:8, Ile:50, 54 | Gly:48, 49, 51, 52, Phe:53, Ile:47, Thr:80, 84, Asp:25, Pro:81, Leu:23, Pro:9, Arg:8 |
| Corotoxigenin | −9.63 | 2 | Asp:29, 30 | Ala:28, Gly:48, 49, Ile:50, Gly:51, 52, Pro:81, Phe:53, Thr:80, Ile:84, 47, Val:32, Ile:54 |
| Calotropogenin | −9.6 | 2 | Asp:29, 30 | Thr:80, Ala:28, Leu:76, Ile:50, Gly:48,49, 51, 52, Pro:81, Ile:47, 54, 84, Val:82 |
| Procesterol | −9.37 | 0 | No hydrogen bonds | Arg:8, Val:82, Pro:81, Thr:80, Val:32, Asp:25, Ile:84, Asp:30, Gly:27, Ala:28, Asp:29, Gly:48, Ile:47, Gly:49, Phe:53, Gly:51,52, Ile:50, 54 |
| 7β,14 Dihydroxy-5α-Card-20(22)-Enolide | −9.35 | 0 | No hydrogen bonds | Thr:80, Gly:52, Phe:53, Pro:81, Val:32, Asp:29, 30, Gly:49, 48, 51, Ile:50, 54, 47, 84, Ala:28 |
| Coroglaucigenin | −9.04 | 3 | Asp:29, 30, Gly:48, Ile:50 | Gly:49, 51, Leu:76, Pro:81, Thr:80, Ala:28, Ile:47, 84, Val:32 |
| Standard (APV) | −6.77 | 3 | Asp:25, Ile:50, Gly:48 | Gly:49, 51, 52 Ala:28, Asp:30, Asp:29, Ile:47, Val:32, Ile:54, Phe:53, Thr:80, Val:82, Ile:84, Pro:81 |
Asp, aspartic acid; Gly, glycine; Thr, threonine; Ala, alanine; Asp, aspartic acid; Gly, glycine; Ile, isoleucine; Phe, phenylalanine; Val, valine; Arg, arginine; Ile, isoleucine; Met, methionine; Pro, proline; Leu, leucine
Furthermore, among the steroidal cardenolides, voruscharin and uscharidin were the top two compounds with binding energies lower than -10 kcal/mol. Voruscharin had the lowest binding energy of -12.38 kcal/mol, followed by uscharidin with a binding energy of -11.32 kcal/mol. This suggested that these two steroidal cardenolides had the strongest binding interactions with HIV-1 PR among the compounds tested. The diagrammatic representation is given in Fig. 1 and 2, in which the ligands (voruscharin and uscharidin) are shown as a ball-and-stick model with hydrogen-bonds represented by green dotted lines; oxygen atoms in red and hydrogen atoms in white. The possibilities of hydrogen bond formation between the compound and amino acids are presented.
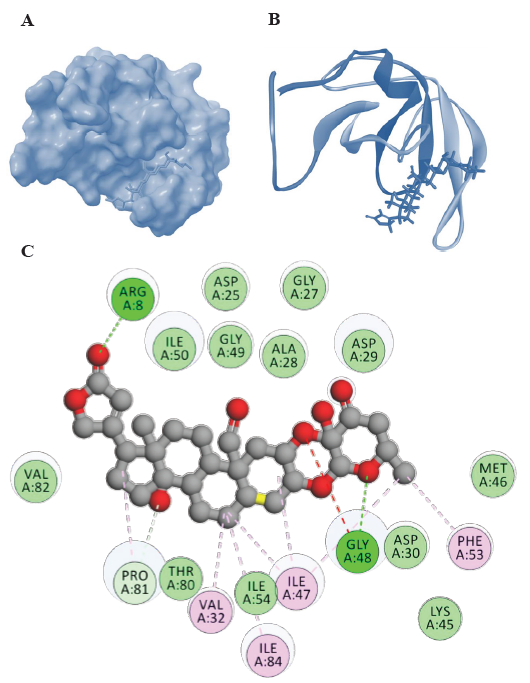
- Representations of HIV-1 PR-Uscharidin complexes from molecular docking visualized using Biovia Discovery Studio visualizer (
https://discover.3ds.com/discovery-studio-visualizer-download). (A) Surface representation of binding confirmation, (B) 3D diagram of binding and (C) 2D diagram of binding conformation of HIV-1 PR with Uscharidin along with the H bonds formed in the complex and hydrophobic interactions between HIV-1 PR and Uscharidin in the docked complex. Lys, lysine.
Binding interactions of voruscharin with HIV-1 PR
The interactions between voruscharin and specific amino acid residues in HIV-1 Protease played an important role in the stability and specificity of molecular interactions in the protease. Voruscharin had the lowest binding energy of -12.38 kcal/mol and these hydrogen bond interactions involved the nitrogen and oxygen atoms of Voruscharin and the amino acid residues Asp29, Thr80, and Gly27 in HIV-1 Protease (Fig. 1). Additionally, the nitrogen atom of Asp29 and the oxygen atom of Thr80 also interacted with the oxygen atom of Voruscharin, further highlighting the potential binding and interactions between Voruscharin and HIV-1 Protease. Voruscharin interacted with other residues such as Ala28, Asp25, 30, Gly48, 49, Ile50, Phe53, Thr80, and Val82 with hydrophobic interactions (Table and Fig. 1). Understanding these interactions can provide valuable insights for the design of potential inhibitors or drugs targeting HIV-1 Protease, which can aid in the development of novel therapeutic strategies against HIV infection.
Binding interactions of uscharidin with HIV-1 PR
Uscharidin had the lowest binding energy of -11.32 kcal/mol and forms two hydrogen bonds with Arg8 and Gly48 (Fig. 2). It also interacted with other residues such as Arg8, Gly27, 48, 49, Val32, 82, Ile47, 50, 84, Phe53, Asp25, 30, Ala28, Asp29, Met46, Thr80, and Pro81 (Table and Fig. 2). The C=O of the uscharidin acted as hydrogen acceptor and formed hydrogen bond interaction with Arg8 of the HIV-1 PR at a distance of 1.97Å. Similarly, the Gly48 residue had a hydrogen bond donor atom, hydrogen, which interacted with the acceptor atom, oxygen, of Uscharidin at a distance of 2.46Å. These findings suggested that Uscharidin could form hydrogen bonds with specific amino acid residues at different distances, indicating potential interactions and stability within the system.
Binding interactions of other steroidal cardenolides with HIV-1 PR
Among the compounds, Beta-Anhydroepidigitoxigenin exhibited hydrogen bond interactions with a binding energy of -10.94 kcal/mol towards Gly48 and Gly27 (Table). This indicated a specific binding affinity towards these residues. Afroside formed hydrogen bond interactions with Asp29 and Gly48, indicating potential binding to these residues. Calactin formed hydrogen bond interactions with Asp29, Gly48, and Thr80, indicating specific binding to these residues. All of the compounds showed interactions with various residues in the active site regarding hydrophobic interactions. These interactions contributed to the overall stability and binding affinity of the compounds.
Comparing the compounds to the standard (APV), it was evident that all of the compounds had a stronger binding affinity towards the active site of HIV-1 Protease. This indicated their potential as inhibitors of the protease activity. The specific molecular interactions observed between the compounds and the active site residues provided valuable insights into their mechanism of action and potential for further development as therapeutic agents. Overall, these findings highlight the significance of natural product compounds in drug discovery efforts targeting HIV-1 Protease.
Molecular dynamic simulation
A computational process was conducted using molecular dynamics (MD) simulation study to evaluate the stability of the protein HIV-1 PR and the top potent docked compound Voruscharin with HIV-1 PR and compared with the standard drug APV. The study aimed to investigate the motion of the complex compound and protein alone at the atomic level by performing a stability analysis using RMSD, RMSF, and Radius of gyration values as a function of time.
The root means square deviation (RMSD)
The RMSD values of the protein HIV-1 PR and protein-ligand complexes were calculated from 0 to 10ns to determine the structure variation. The RMSD values steadily increased from 0 to 2ns and reached a stable state throughout the simulation. However, fluctuations were observed in the lone HIV-1 PR structure between 6 to 8ns. The RMSD plots (Fig. 3) for the complex (HIV-1 PR-Voruscharin) indicated that the compound showed lower fluctuations than the protein alone within the simulation time. Additionally, the RMSD value of HIV-1-PR complex with Voruscharin was lower (0-0.3nm) compared to HIV-1-PR alone (0-0.4nm), indicating the stability of HIV-1 PR when bound to ligand voruscharin. The RMSD value of HIV-1-PR complex with standard drug APV was also similar when compared to HIV-1-PR complex with voruscharin. Therefore, the RMSD analysis suggested that the binding of Voruscharin to HIV-1 PR enhanced its stability.
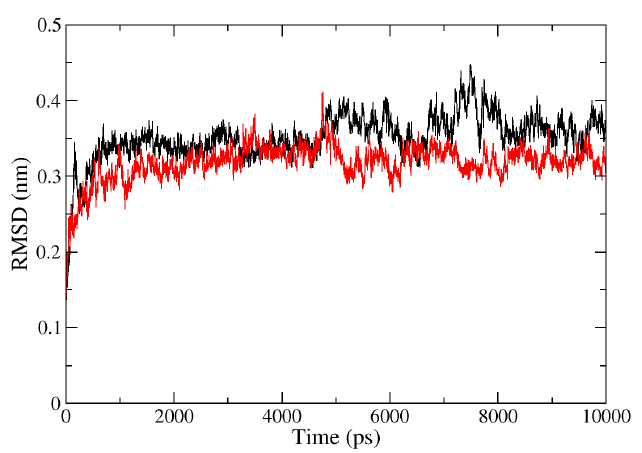
- Representation of MD simulation trajectory RMSD plot of the protein bound to the ligands visualized using QtGrace software. The RMSD of HIV-1 PR with Voruscharin (red) and HIV-1 PR alone (black) were obtained during 10000 ps (10 ns) simulation. RMSD, root mean square deviation; MD, molecular dynamics.
Root means square fluctuation (RMSF)
RMSF analysis provided information about the fluctuation of each atom in the residues during the MD simulation of the lone protein HIV-PR and the protein-ligand complex HIV-1 PR with voruscharin (Fig. 4). For the HIV-1 PR with voruscharin structure, RMSF was calculated using 888 atoms of 99 amino acids and one ligand voruscharin. The results confirmed that the binding site residues, specifically Asp29, Gly27, and Thr80 showed less fluctuations. Higher RMSF values indicated greater flexibility during the MD simulation. Compared to the lone HIV-PR structure, the RMSF values for regions 27-29 significantly increased for the HIV-1 PR with voruscharin structure, with a value of 0.13 nm for the alone HIV-1 PR structure. There were no significant changes observed in the RMSF values of HIV-1 PR with Voruscharin when compared to HIV-1 PR with the standard drug APV. These larger RMSF values indicated increased random motions of these residues, particularly at the binding residue sites of Gly27 and Asp29. This suggested that the binding of Voruscharin to HIV-1 PR introduced additional flexibility in these regions.
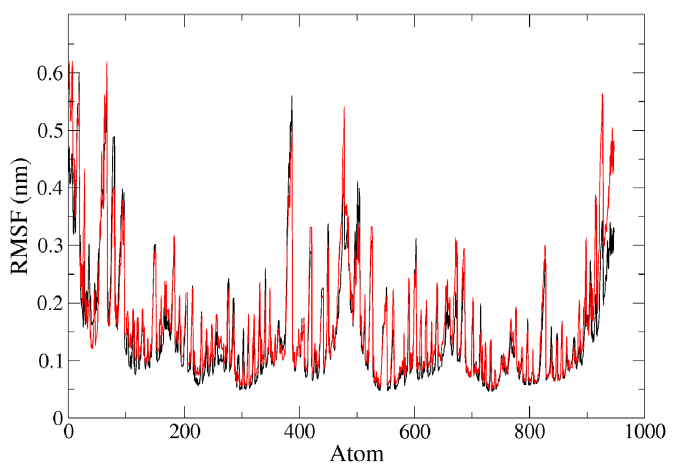
- Representation of MD simulation trajectory RMSF plot of the protein bound to the ligands visualized using QtGrace software. The RMSF of HIV-1 PR with voruscharin complex (red) and HIV-1PR alone (black) were obtained during 1000 ps (10 ns) simulation. RMS, root mean square; RMSF, root mean square fluctuation.
Radius of gyration (Rg)
The radius of gyration is a significant parameter that can be used to investigate the motion and stability of a protein by describing its compactness during the simulation time. The initial radius of gyration (Rg) values of the protein and ligand complexes were observed to be between 1.32 and 1.41 nm (Fig. 5). During the simulation, the Rg value of HIV-1 PR with Voruscharin was observed to be higher compared to HIV-1 PR alone. The initial Rg value was higher than that at the end of the simulation, indicating the stability of the HIV-1 protein complexed with Voruscharin throughout the simulation period. This suggested that the protein maintained a stable conformation in the presence of Voruscharin, reflecting the effective binding interactions between the compound and the HIV-1 protease. When comparing the Rg values of HIV-1 PR with the standard APV and with Voruscharin, they showed more stabilization after 2 ns. In contrast, for HIV-1 PR alone, the values initially decreased and stabilized from 3 to 8 ns, indicating a stable binding pose. Therefore, the radius of gyration is an interesting parameter that can provide valuable insights into the stability of the protein-ligand complex during MD simulations.
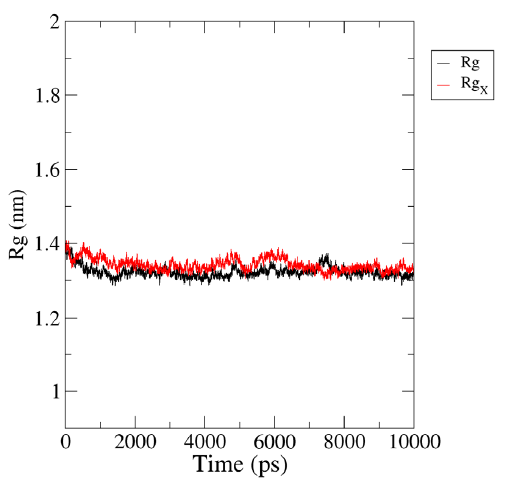
- Representation of MD simulation trajectory Rg plot of the protein bound to the ligands visualized using QtGrace software. The Rg of HIV-1 PR with Voruscharin complex (red:Rgx) and HIV-1 PR alone (black:Rg) were obtained during 1000 ps (10 ns) simulation. Rg, radius of gyration; Rgx, radius of gyration for rotation around the X axis.
Discussion
The binding energies obtained from the analysis provide a measure of the strength of the ligand-receptor interaction, with lower values indicating stronger binding affinity13. The results showed that all of the steroidal cardenolides exhibited higher binding affinity than APV, indicating their potential as inhibitors of HIV-1 PR. The binding energies of the steroidal cardenolides were significantly lower than that of APV (-12.38 to -9.04 kcal/mol), suggesting a higher affinity for their target receptors.
Hydrogen bond interactions serve as a measure of the quantity and nature of hydrogen bonds established between a ligand and its target receptor. A greater number of hydrogen bond interactions signifies a more robust binding interaction14. In the present study, Voruscharin formed three hydrogen bonds with the Asp29, Thr80, and Gly27 residues of HIV-1 PR, while Uscharidin engaged in two hydrogen bonds with the Arg8 and Gly48 residues of the same protein. These interactions further support the binding affinity of Voruscharin and Uscharidin towards HIV-1 PR, suggesting its potential as a promising compound for inhibiting protease activity.
The development of antiretroviral drugs has targeted the active site of HIV-1 PR, which has proven to be a well-defined target. Inhibitors designed to bind to this active site have successfully blocked the proteolytic activity of HIV-1 PR, leading to the development of effective antiretroviral therapies15. The active site of HIV-1 PR consists of two aspartic acid residues (Asp25 and Asp29) and is located within a cleft between two identical subunits of the protein16.
The active site interactions of HIV-1 Protease (PDB ID: 3NU3) involved several key residues (Asp25, Asp29, Glu35, Gly27, Gly48, Ile47, Ile50, Ile54, Lys45, Val32) that played a crucial role in its catalytic activity17. In this study, the compounds Voruscharin and Uscharidin interacted with multiple active residues, including Asp25 and Asp29.Voruscharin also interacted with Glu35, Gly27, Gly48, Ile50, and Val32 and Uscharidin interacted with Gly27, Gly48, Ile47, Ile50, and Val32 (Fig. 3). These interactions are important for substrate binding and cleavage, contributing to the overall function and specificity of the protease18. Understanding these active site interactions can provide valuable insights for the design of potential inhibitors or drugs targeting HIV-1 Protease.
Nukoolkarn et al19 conducted 20 ns MD simulations of main protease and inhibitor complexes (ritonavir and lopinavir) and found that the ligand-binding site of the protease was located around the active sites of H41 and C145. RMSD is a numerical measurement used to estimate the approximate distance between a group of atoms, mainly the backbone atoms of a protein, plotted against time. This measurement provides an indication of how much the protein’s structure has been modified over time in comparison to its starting point. The value of RMSD is inversely correlated with the stability of the backbone atoms; therefore, a higher RMSD value (>2nm) indicates less stability in the backbone atoms20. This information was documented by Mamidala et al21 in their study on RMSD and its applications in protein structure analysis. In the same study, the RMSD values initially increased (<2nm) and then reached a stable state, suggesting a stable conformation of the protein-ligand complexes.
RMSF was utilized in our study to examine the impact of amino acid mutations on the conformational flexibility of HIV-1 PR, both in its unbound form and when bound to a ligand. It is generally considered acceptable for small proteins to have RMSF fluctuations below 2Å, indicating relatively stable conformations22. Our studies showed that HIV-1 PR alone decreased RMSF values for specific residues, indicating reduced flexibility and enhanced stability in those regions. Additionally, we utilised the Rg calculated from the MD trajectory to assess the compactness and rigidity of the protein system during the simulation. Higher Rg values suggest a less compact protein structure, indicating greater flexibility. Conversely, lower Rg values indicate higher stability and compactness of the protein structure. The Rg analysis performed in this study, with Rg values ranging from 1.5nm to 2.5 nm, was consistent with the findings of Khan et al23 who reported that higher Rg values indicated less compactness and lower stability of protein structures. Conversely, lower Rg values suggested greater stability and compactness.
Overall, based on the molecular docking study, it appears that steroidal cardenolides derived from C. procera have potential as inhibitors of HIV-1 protease. The binding energy measurements suggested that these compounds might have a higher affinity for the HIV-1 protease receptor than the standard drug, Amprenavir. Specifically, the steroidal cardenolides Voruscharin and Uscharidin, demonstrated the strongest binding interactions. The RMSD of complex fall and high RMSF values inferred that the compound Voruscharin had undergone good conformational changes while binding, and maintained close affinity with the binding site of the HIV-1 PR. Given these results, it seems that Calotropis procera could potentially offer a source of compounds for developing novel HIV-1 protease inhibitors. Further studies are required to assess the efficacy of these compounds against HIV-1 PR with cell-based assays and enzymatic kit to validate these findings and also clinical trials are necessary to evaluate their safety and potential as a drug candidate for treating HIV-1 infection.
Financial support & sponsorship
The study was financially supported by the grant (HIV/fellowship/4/10/2020-ECD-11) received from the Indian Council of Medical Research, New Delhi, India to first author (KS).
Conflicts of Interest
None.
Use of Artificial Intelligence (AI)-Assisted Technology for manuscript preparation
The authors confirm that there was no use of AI-assisted technology for assisting in the writing of the manuscript and no images were manipulated using AI.
References
- HIV/AIDS, 2021. Available from: https://www.who.int/news-room/fact-sheets/detail/hiv-aids, accessed on October 1, 2023.
- Current status of antiretroviral therapy. Clin Epidemiol Glob Health. 2019;7:532-7.
- [Google Scholar]
- Natural products as HIV-1 protease inhibitors: A review. Phytother Res. 2021;35:532-7.
- [Google Scholar]
- Antiviral activity of calotropis procera against HIV and herpes simplex virus. Indian J Pharmacol. 2020;52:357-61.
- [Google Scholar]
- Identification of potential natural compounds as HIV-1 protease inhibitors using molecular docking and simulation studies. J Biomol Str Dyn 2021:1-14.
- [Google Scholar]
- Antiviral activity of 3-O-methylfunicone isolated from aspergillus fumigatus against HIV-1. J King Saud Univ Sci. 2021;33:101290.
- [Google Scholar]
- Herbal medicine: An effective therapeutic approach for the management and prevention of HIV/AIDS. Curr Pharm Des. 2019;25:2927-33.
- [Google Scholar]
- HIV-1 protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS (Auckl). 2020;12:299-308.
- [Google Scholar]
- RCSB protein data bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2018;47:464-74.
- [Google Scholar]
- AutoDock4 and autodocktools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785-91.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Pharmacophore screening, molecular docking, ADMET prediction and MD simulations for identification of ALK and MEK potential dual inhibitors. J Mol Struct. 2021;1245:131066.
- [CrossRef] [Google Scholar]
- Phenolic compounds as promising drug candidates against COVID-19-an integrated molecular docking and dynamics simulation study. Mater Today Proc. 2022;51:522-7.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The statistical-thermodynamic basis for computation of binding affinities: A critical review. Biophys J. 1997;72:1047-69.
- [CrossRef] [PubMed] [Google Scholar]
- Rilpivirine inhibits SARS-CoV-2 protein targets: A potential multi-target drug. J Infect Public Health. 2021;14:1454-60.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- HIV protease inhibitors activate the unfolded protein response and disrupt lipid metabolism in primary hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2006;291:1071-80.
- [CrossRef] [Google Scholar]
- Molecular docking and molecular dynamics studies on β-lactamases and penicillin binding proteins. Mol Biosyst. 2014;10:891-900.
- [Google Scholar]
- Amprenavir complexes with HIV-1 protease and its drug-resistant mutants altering hydrophobic clusters. FEBS J. 2010;277:3699-714.
- [Google Scholar]
- Oral absorption of the HIV protease inhibitors: A current update. Adv Drug Deliv Rev. 1999;39:211-38.
- [CrossRef] [Google Scholar]
- Molecular dynamic simulations analysis of ritronavir and lopinavir as SARS-CoV 3CLpro inhibitors. J Theor Biol. 2008;254:861-7.
- [CrossRef] [Google Scholar]
- Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys J. 2010;98:861-71.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- In silico prediction of mozenavir as a potential drug for SARS-CoV-2 infection via binding multiple drug targets. Saudi J Biol Sci. 2022;29:840-7.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J Biomol Struct Dyn. 2021;39:2607-16.
- [CrossRef] [PubMed] [Google Scholar]
- Effect of flap mutations on structure of HIV-1 protease and inhibition by saquinavir and darunavir. J Mol Biol. 2018;381:102-15.
- [Google Scholar]




