
Translate this page into:
Insights into existing and futuristic treatment approach for chronic myeloid leukaemia
For correspondence: Dr Hemant Ritturaj Kushwaha, School of Biotechnology, Jawaharlal Nehru University, New Delhi, 110 067, India e-mail: hemantkushwaha@jnu.ac.in
-
Received: ,
Abstract
Oncogenes play a crucial part in human cancer development, and when particular drugs obstruct the proteins produced by these oncogenes, the tumoural process can be ceased. For instance, in chronic myeloid leukaemia (CML), all pathological traits are associated with a single oncogene, BCR-ABL1. CML is a triphasic cancerous disorder of haematopoietic stem cells, marked by a balanced translocation between chromosomes 9 and 22, leading to the genesis of a Philadelphia chromosome encompassing the BCR-ABL1 fusion gene. This fusion oncogene further produces a constitutive active tyrosine kinase protein, enhancing the downstream signalling pathways and constitutes cancer. The treatment for CML has been entirely altered from chemotherapy and immunotherapy to targeted therapy with the emergence of tyrosine kinase inhibitors (TKIs) which inhibit BCR-ABL1 kinase activity. However, the inhibitory mechanism of TKIs is constrained by BCR-ABL1 dependent and independent resistance mechanisms, prompting the exploration of novel therapeutics through extensive clinical trials to develop next-generation drugs with enhanced potency. The persistent challenges posed by CML have motivated researchers to seek innovative strategies for its eradication, such as the application of the genome editing tool CRISPR/Cas9. This review provides insights into existing CML diagnoses, treatment modalities, resistance mechanisms, drugs under trial phases and new potential therapeutic drugs. Furthermore, the review looks ahead to a visionary perspective wherein the CRISPR/Cas9 approach holds the potential to evolve into a prospective curative measure for CML.
Keywords
Chronic myeloid leukaemia
clinical trial
CRISPR/Cas
drug resistance
treatment-free resistance (TFR)
tyrosine kinase inhibitors (TKIs)

Cancer is a rising problem worldwide, with around 19.3 million new cancer cases in addition to 10 million cancer deaths being reported in 20201. Among all 36 types of cancers, leukaemia is the 11th globally (Global Cancer Statistics, 2020)1. Under leukaemia, the bone marrow makes an enormous number of aberrant white blood cells (WBCs) that are not entirely developed and are known as blasts. Generally, leukaemia alters the leukocytes or WBCs. One of the existing types of leukaemia is chronic myeloid leukaemia (CML). CML, a non-hereditary disease, typically occurs in middle-aged individuals and accounts for 15-20 per cent of all leukaemia2. CML develops as a result of chromosomal translocation of the ABELSON 1 gene (ABL1, 171.74 kb) on chromosome 9 to the Breakpoint Cluster Region gene (BCR, 137.83 kb) on chromosome 22, resulting in shorter chromosome 22 - Philadelphia (Ph) chromosome [t(9,22) (q34.1,q11.2)] as shown in Figure 13. The Philadelphia chromosome is present in 90 per cent of CML cases. BCR-ABL gene acquisition initially occurs in a single haematopoietic stem cell (HSC) that attains high proliferation capacity, giving rise to an increased number of blast cells. According to WHO 2016 criteria, CML has three different stages of progression initiating from the chronic phase (CML-CP) to advanced stages like accelerated phase (CML-AP) and blast phase (CML-BP). Acquisition of new chromosomal aberrations in Ph (+) cells known as additional chromosomal abnormalities (ACA), is one of the main factors of progression from CML-CP to CML-AP and CML-BP4. CML influences both sexes, with a 2.2 male to 1.4 female ratio5. The Global Burden of Disease (GBD) study 2019 shows that CML incidence cases were raised from 1990 to 2019 by 54.1 per cent, while the global death rates decreased slightly during this period6. The CML burden differs in different countries because of the difference in the availability of premature screening methods, novel drugs and medical equipment.
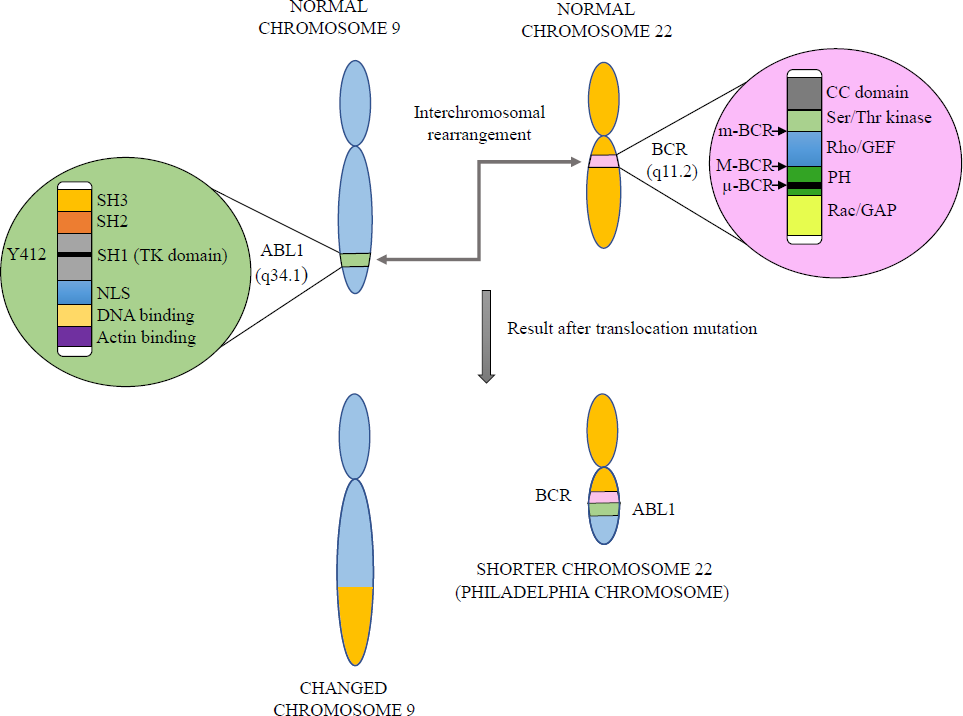
- Chromosomal translocation process in CML. Source: Ref 3.
It is well known that cancer is one of the most common human genetic disorders where alteration from a normal cell to a malignant cell is governed by modifications to a cell’s DNA, termed mutations. The reasons leading to the DNA mutation and making it cancerous are still unresolved. However, some risk factors which may elevate the chances of acquiring CML include smoking, high body mass index (BMI) and obesity, radiation exposure, occupational exposure, age and gender. In third-world countries, smoking is one of the crucial contributors to CML6.
Symptoms of CML comprise weakness, tiredness, weight loss, high fever, anaemia, bone pain, enlarged spleen and a sense of fullness in the belly. However, similar signs can also be present in other diseases. CML does not have any particular symptoms, but there are specific methods of diagnosis and prognosis. The contemporary landscape of CML therapy is characterised by a diverse range of treatment modalities. Predominantly, targeted therapy facilitated through tyrosine kinase inhibitors (TKIs) has emerged as a focal point in CML treatment, featuring first, second and third-generation TKIs with substantial potency. The molecular assessment of BCR-ABL transcript levels for monitoring treatment effectiveness is crucial, a determinant factor in achieving treatment-free remission (TFR). However, in spite of these advances, the persistence of the CML burden remains attributable to mechanisms of resistance that undermine the sustained success of therapeutic interventions. Given the urgency of this challenge, there exists a critical need to formulate innovative strategies that not only address the primary drivers of the disease but also confront the intricate network of resistance mechanisms. This review delivers a comprehensive literature survey encompassing all facets of CML and its potential cure.
Methods
A laboratory blood test, complete blood count (CBC), a well known laboratory blood test is the initial step in the diagnosis of CML (Leukemia and Lymphoma Society). The next stage of diagnosis is bone marrow aspiration and biopsy, where blood is collected for cytogenetic and molecular examinations. For performing diagnosis, it is mandatory to reveal the existence of the Ph chromosome through karyotype examination or detection of the BCR-ABL transcript by reverse transcriptase-polymerase chain reaction (RT-PCR) and fluorescence insitu hybridisation (FISH)7. In some cases, mutations occur in the BCR-ABL1 gene and this leads to false results for other diagnostic tests. Next-generation sequencing (NGS) and Sanger sequencing are typically used in such cases of mutation with, NGS having a better limit of detection (LOD)8. LOD is the lowest concentration or amount of the substance being analysed in a sample that can be precisely distinguished from zero. Table I represents comparative details of all the existing diagnostic methods in CML with their respective targets, LOD, sample type, turnaround time, advantages, disadvantages and costs (in range as quoted by different manufacturers)8-12.
| Diagnosis Methods | Target |
Limit of Detection (I.S %) |
Type of Sample |
Turn Around Time (days) |
Application | Advantages | Disadvantages |
Cost (Range) |
References |
|---|---|---|---|---|---|---|---|---|---|
|
CBA/ Karyotyping |
Philadelphia chromosome, t (9;22) | 5 | Blood from bone marrow (BM) | 3 | Diagnosis |
No earlier knowledge needed Overview includes all Chromosomal aberrations |
Necessity of dividing cells Time consuming Limited resolution (Mbp) Occasionally complex karyotypes can lead to false identification of t (9;22) |
Rs. 3200 – Rs. 4000 | 8, 11, 12 |
| FISH | BCR – ABL1 DNA | 0.1-5 | Peripheral blood (PB) and bone marrow (BM) | 2 |
Diagnosis, Quantification of major breakpoint cluster region |
Can identify variation that are minute to be seen under a microscope No obligation of vital cells |
No identification of additional aberrations Only BCR-ABL detectable Insensitive in contrast to RT-PCR |
Rs. 5000 – Rs. 8000 | 8, 10, 11, 12 |
|
RT-qPCR (Old method) |
BCR – ABL1 mRNA (fusion transcript) | 0.001-0.01 | Peripheral blood (PB) | 1 |
Quantify BCR – ABL1 transcript levels, Determine breakpoint of fusion gene |
Very sensitive Determine breakpoints of fusion gene Widely available |
Cross contamination Necessity of standard curve Sensitive to inhibitors |
Rs. 6000 – Rs. 8000 | 8, 10, 11, 12 |
|
digital PCR (d-PCR) (New method) |
BCR – ABL1 mRNA (fusion transcript) or cDNA | 0.0001-0.001 | Peripheral blood (PB) | 2 |
Quantify BCR – ABL1 transcript levels |
More sensitive, economical and error-free Facilitate the observation of as little as 1 copy of BCR-ABL1 target |
Not yet broadly accessible Not yet standardized Can be executed only for a restricted number of mutations |
Rs. 9000 – Rs. 11,000 | 8, 9, 11 |
|
Sanger sequencing (Old method) |
Kinase Domain of BCR – ABL1 gene | 20 | RNA or DNA from peripheral blood buffy coat | 6 | BCR – ABL1 Kinase Domain mutation testing |
Widely accessible Easy to operate |
Poor sensitivity | Rs. 5000 – Rs. 10,000 | 9,10 |
|
NGS (New method) |
Kinase Domain of BCR – ABL1 gene | 1-3 | RNA or DNA from peripheral blood buffy coat | 11 | BCR – ABL1 Kinase Domain mutation testing |
More sensitive than Sanger sequencing Permits scanning of whole Kinase domain for any mutation |
Labor intensive Not yet broadly accessible To be economical, demands pooling of 8-10 samples |
Rs. 15,000 – Rs. 48,000 | 9,11 |
BM, bone marrow; CBA, chromosome banding analysis; dPCR, digital polymerase chain reaction; FISH, fluorescence in-Situ hybridisation; I.S, international standard; NGS, next generation sequencing; PB, peripheral blood; RT-qPCR, reverse transcriptase-quantitative polymerase chain reaction; RT-PCR, reverse transcriptase-polymerase chain reaction
| First-line treatment | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Generation | Drug (Generic name) | Trade name | Standard dose | International randomized trials | Side effects | References | |||||
| Trial name | Intervention | Patients | Response | ||||||||
| First generation TKI |
Imatinib Mesylate (IM) (STI571) |
Gleevec |
400 mg/day |
IRIS | Imatinib (400mg) | 1106 |
At 1 yr- MMR: 20-59 |
At 5 yr- MMR: 60-80% PFS: 80-90% OS: 90-95% |
At 11 yr- MMR: 93% CCyR: 83% OS: 83.3% |
Blood in urine, eye and nose, abdominal or stomach pain, cramping, burning, or tenderness | 13 |
| Second generation TKI (2GTKI) | Dasatinib | Sprycel |
100 mg/day |
DASISION |
Dasatinib (100mg) (1) vs. Imatinib (400mg) (2) |
519 |
At 2 yr- CCyR: 86% (1) 82% (2) |
At 5 yr- MMR: 76% (1) 64% (2) PFS: 85% (1) 86% (2) OS: 91% (1) 90% (2) |
Anemia, neutropenia, thrombocytopenia, bleeding, rash and headache | 14 | |
|
Nilotinib (NIL) |
Tasigna |
300mg twice a day |
ENESTnd |
NIL 400mg (1) NIL 300mg (2) IM 400mg (3) |
846 |
At 2 yr- CCyR: 87% (1) 85% (2) 77% (3) |
At 5 yr- MMR: 77% (1) PFS: 95% (1) 77 %- 97%(2) 60% - 93%(3) |
At 6 yr- OS: 92% (1) 96% (2) 91% (3) |
Familiar side effects - Low blood count, nausea, rash, headache Severe side effects- heart and blood vessel complication |
15 | |
| Bosutinib | Bosulif |
400 mg/day |
BFORE |
Bosutinib (400mg) (1) vs. Imatinib (400mg) (2) |
536 |
At 1 yr- MMR: 47% (1) 37% (2) CCyR: 77% (1) 66% (2) PFS: 96% (1) 94% (2) OS: 99.9% (1) 99.7% (2) |
Diarrhea, nausea, vomiting, rash, indication of pancreatitis, and rise of serum lipase | 16 | |||
| Radotinib | Suspect |
Not approved by FDA, only approved in South Korea. |
RERISE |
Radotinib (300mg twice a day) (1) vs. Imatinib (400mg/day) (2) |
241 |
At 1 yr- MMR: 52% (1) 30% (2) CCyR: 91% (1) 77% (2) |
At 4 yr- MMR: 86% (1) 75% (2) OS: 99% (1) 94% (2) PFS: 97% (1) 94% (2) |
Severe or even life-threatening coronary artery disease, QT prolongation, changes in left ventricular ejection fraction |
17 | ||
| Second-line treatment | |||||||||||
| Third generation TKI | Ponatinib | Iclusig |
45 mg/day |
PACE | Ponatinib | 449 |
At 1 yr- MCyR: 56% |
At 5 yr- MCyR: 82% OS: 73% |
Abdominal pain, headache, dry skin, rash and constipation |
18 | |
BFORE, bosutinib trial in first-line chronic myeloid leukaemia treatment; CCyR, complete cytogenetic response ((BCR-ABL ≤ 1% International Scale); DASISION, DASatinib versus imatinib study in treatment-naive chronic myeloid leukaemia patients; ENESTnd, evaluating nilotinib efficacy and safety in clinical trials-newly diagnosed patients; IRIS, international randomized study of interferon and ST1571; MCyR, major cytogenetic response (less than 35% of the cells in the bone marrow have the Ph chromosome); MMR, major molecular response (BCR-ABL ≤ 0.1% International Scale); OS, overall survival; PACE, ponatinib ph-positive acute lymphoblastic leukaemia (ALL) and CML evaluation; PFS, progression-free survival; RERISE, randomized evaluation of radotinib versus imatinib for efficacy
Treatment
CML treatment relies on the disease phase at the time of diagnosis. Also, one patient can receive either only one type of therapy or a combination of therapies. Initially, therapeutic modalities for CML encompassed chemotherapeutic agents and immunomodulatory approaches; however, since the advent of tyrosine kinase inhibitors (TKIs) in 2000 for targeted therapy, this class of pharmaceuticals has substantially supplanted conventional treatment strategies.
Targeted therapy
In CML, TKI drugs target the BCR-ABL kinase enzyme. There are only five TKIs accessible for treating CML which are divided as first and second line treatment.
First-line treatment and second-line treatment
First-line treatment comprises TKIs for newly identified CML patients. Presently, only four TKIs are finalised under firstline treatment against CML. This includes first-generation drug imatinib and second-generation drugs (2GTKIs) like dasatinib, nilotinib and bosutinib. All these TKIs have similar working mechanisms. These drugs bind to ATP binding sites on the kinase domain of BCR-ABL oncoprotein and then obstruct the transfer of the phosphate group to the protein substrate and its consecutive stimulation. As a result, proliferative signals are blocked, and apoptosis is induced in leukaemic cells. The details of all these TKIs are mentioned in Table II13-18. Firstline treatment is shifted to secondline treatment after its failure, i.e. resistance or intolerance of initial TKI. The sole TKI which cannot be used in firstline treatment and is developed only for secondline treatment is ponatinib. Failure of ponatinib after three months of treatment leads to the recommendation of early allogeneic-stem cell transplantation (Allo-SCT).
Treatment beyond second-line: stem cell transplantation (SCT)
An insignificant reaction to two or more TKIs leads to considering SCT. The objective of SCT is to eradicate cancer cells in the body either by chemotherapy or by radiation and then substitute the bone marrow containing leukaemic cells with blood-forming HSCs. Out of autogenic and allogenic, exclusively allogenic-SCT (Allo-SCT) is used to heal CML in which healthy blood stem cells are taken from another person of the same species and then transplanted into the diseased patient. In advanced CML, second or third-generation TKIs are introduced to bring down CML load and then recommended for premature Allo-SCT19,20. If primary 2GTKI prescribed under first or second-line treatment fails, the patient is recommended Ponatinib or an experimental agent. Simultaneously, the patient is evaluated for Allo-SCT and a donor hunt begins. Patients in CML-AP should be considered high-risk patients and should proceed to SCT without a lag as progression-free survival (PFS) in the blast phase is extremely low.
Molecular monitoring & treatment-free remission (TFR)
Molecular monitoring of BCR-ABL1 transcripts is used to evaluate the response of TKIs in CML patients. It is used to check the treatment failure and to alter the therapy timely. Although the clear-cut BCR-ABL1 transcript level and duration of the maintenance period after which treatment termination can be tried have not yet been decided, it is expected that the results of clinical trials on the TKI termination study will be out in the coming years. Presently, major molecular response (MMR, BCR-ABL1 ≤ 0.1% on International Standard (IS)) is an ideal response. Further deep molecular response (DMR) is a new frontier in CML treatment. DMR can be described as BCR-ABL1 levels of molecular response (MR)⁴ (BCR-ABL ≤ 0.01%) and molecular response (MR)⁴,⁵ (BCR-ABL ≤ 0.0032%) on the IS21. Non-success in obtaining MMR by the 12 months is related to a low rate of deep response22. Hence, molecular monitoring is an essential element in managing patients with CML and the TFR of recovered patients.
Treatment-free remission in medical terms refers to a decrease in or disappearance of signs and symptoms of cancer for a specific period. However, only 40-60 per cent of patients showed accomplishment in attaining TFR23. The first investigation on the concept of ending TKI treatment was through the Stop Imatinib 1 (STIM1) study24 in which 100 patients who attained complete molecular response (CMR) after more than two years of IM treatment were terminated from TKI therapy. Further, 42 patients (61%) experienced molecular relapse after TKI therapy termination, while 26 patients again attained CMR after IM reintroduction. Whereas, after stopping nilotinib and dasatinib, the chance of upholding TFR has been analogous to the results after stopping IM (approximately 50%)25,26. More extended periods of TKI therapy, DMR and former treatment with IFN-α were spotted as crucial prognostic factors for TFR success in the EURO-SKI trial27, the largest trial of TKI discontinuation After support from ENESTfreedom (192 wks, phase 2 trial)28 and the ENESTop (192 wks) study, Nilotinib (NIL) has obtained approval from the health authorities to support TFR28,29. In ENESTfreedom and ENESTop studies, the TFR rate measured at the end of the study was 44.2 and 46.0 per cent, respectively. High TFR rates can be accomplished with 2GTKIs due to intense and more sustained MR attained with 2GTKI than IM.
Given that 50-60 per cent of patients with undetectable DMR are anticipated to lose MMR, real-time qPCR may not be the best method for molecular monitoring during TFR30,31. Therefore, enhanced detection methods are needed to boost the perfection in detecting BCR-ABL1 transcripts and further aid in picking up the patients suitable for TFR. In recent years, digital PCR (dPCR) has completely changed how MRD in haematological diseases is molecularly monitored32. In dPCR, the biological sample is divided into numerous distinct reactions, each undergoing more efficient amplification in microscopic partitions. Partition positivity or negativity depends on template molecule presence, and Poisson’s statistics are applied to determine nucleic acid copies in the initial sample. In this manner, a more sensitive absolute quantification can be carried out without the use of an external calibration or standard curve33. dPCR can detect one BCR-ABL1 positive cell in 107 cells and is less susceptible to non-specific amplification and inhibitory agents that can damage DNA34. While dPCR offers advantages and applications, it has limitations, such as longer experimental durations and susceptibility to errors in pre-analytical stages like sampling, RNA extraction followed by cDNA synthesis. When analyzing PB samples directly, the Cepheid GeneXpert system, a cartridge-based automated real-time qPCR technique, can be used to find BCR-ABL1 p210 transcripts. The GeneXpert instrument combines RNA extraction, RT-PCR and BCR-ABL1 fluorescence detection in one reaction by employing microfluidics in a cartridge35. Therefore, this instrument offers the advantage of a quick and easy-to-use method, requiring little technical knowledge. But for an accurate estimation of BCR-ABL1 on the IS, it is imperative to create and use a specific conversion factor (CF), along with calibration specific to the cartridge lot36.
Continuous burden of CML
Massive research has been done in the field of CML, and several treatment modalities and diagnoses have been developed; still, it is a rising global burden. There are two main factors responsible for the loss of response and continuous global burden in CML, i.e. BCR-ABL1 independent and BCR-ABL1 dependent mechanisms of resistance.
BCR-ABL1 independent mechanism of resistance
Drug transporters
Drug transporter proteins mediate drug movement, with balanced TKI influx and efflux crucial for BCR-ABL1 inhibition, while an imbalance contributes to TKI resistance in CML. The human organic cation transporter 1 (OCT1), the membrane influx pump, is highly responsible for IM uptake inside the cell and the ATP-binding cassette (ABC) efflux pump is used for IM movement outside the cells37. Low OCT1 expression is a typical feature of multidrug resistance and is associated with sub-optimal responses. While OCT1 functional activity in leukaemic cells at diagnosis can predict TKI response, the relationship between OCT1 expression and IM transport or response remains controversial, with many studies showing no significant connection38,39. Other transporters, including OCTN2, OATPs and MATE1, are mediators of TKI transport. According to Alves et al.’s observation, OCT1 and OCTN2 expression decreased simultaneously, demonstrating the involvement of multiple influx transporters in the resistance process40. Similarly, overexpression of the ABCB1 gene that encodes P-glycoprotein (P-gp), one of the ABC efflux transporters, could decrease intracellular IM levels, reducing therapeutic efficacy41. The ABCG2 gene encodes the breast cancer resistance protein (BCRP), a key TKI resistance transporter, especially relevant in protecting LSCs42. Reduced drug influx or increased efflux may foster BCR-ABL1 mutations in CML cells and other resistance mechanisms.
Signalling pathways and regulatory factors
In response to BCR-ABL1 inhibition, CML cells can activate alternative signalling pathways like JAK/STAT, PI3K/AKT, RAS/MAPK and SRC, enabling proliferation and survival despite effective BCR-ABL1 suppression43. JAK2 activation by cytokines from cancer and bone marrow niche cells phosphorylates STAT members, including STAT5, which promotes CML development by enhancing the cell cycle and ROS production, inhibiting apoptosis and increasing P-gp expression44. Upon PI3K activation, AKT undergoes phosphorylation and affects several downstream proteins. AKT targets include BAD, where it suppresses apoptotic signal. Additionally, AKT phosphorylates transcription factors FOXO, blocking their activity, thus preventing apoptosis and promoting the cell cycle45. Ma et al46 identified increased RAS/MAPK pathway activity contributing to IM resistance in CML-LSCs. Elevated activity of the protein kinase C (PKC) family within this pathway promotes leukaemia cell proliferation and inhibits apoptosis even without BCR-ABL1 kinase activity. Overexpression of SRC family members like LYN and HCK has been associated with TKI resistance in CML37. These SRC proteins trigger STAT5 activation to promote proliferation and AKT activation to promote survival. Utilising alternative signalling pathways alongside BCR-ABL1 drugs promises to enhance drug response and prevent resistance in CML treatment.
Epigenetic alterations
The BCR-ABL1 mutation not only causes the HSC to become an LSC but also triggers epigenetic reprogramming. The epigenetic processes are generally categorised into three main groups: histones and their modifications, DNA methylation and non-coding RNAs.
Post-translational modifications on histone tails, such as methylation or acetylation, alter chromatin structure and recruit factors. Dysregulated histone-marking systems in CML impact leukaemic cell survival pathways. Polycomb-group (PcG) proteins, including PRC1 and PRC2, are epigenetic regulators dysregulated in CML LSC. Elevated EZH2 in CP-CML LSCs perform tri-methylation of histone H3 on lysine 27 (H3K27me3) at PRC2 target genes, altering CML LSCs survival dependence on EZH247. BMI1, a crucial factor in PRC1 activity, exhibits elevated levels in CD34+ cells with CP-CML and correlates with disease severity. Increased BMI1 expression is associated with decreased CCNG2 (cyclin G2) expression, which inhibits autophagy. Histone variant, γ-H2AX, at DNA damage sites, mediates critical cellular decisions for DNA repair or apoptosis, with dysregulation evident in CML48. In CD34+ CML cells, SIRT1, a NAD-dependent HDAC, is upregulated and has been linked to the survival of LSCs by carrying out deacetylation on a number of targets including p5349. The protein arginine methyltransferases (PRMT) family of HMTs can either activate or inhibit transcription through the methylation of histone arginine. It is believed that PRMT5 plays a crucial role in the epigenetic regulation of canonical Wnt signalling, a pathway that is crucial for LSC function50. Inhibiting PRMT5 activity in CML CD34+ cells decreased LSC numbers invitro51. HDAC inhibitors like Panobinostat, MAKV and Chidamide with IM demonstrated synergistic anticancer responses and increased therapeutic effectiveness in CML cells52.
Transcription of genes is inhibited by DNA methylation. Proliferation and motility of CML LSC and progenitor cells are likely to be increased by MTSS1 (a tumour suppressor) repression but can be decreased by MTSS1 enforced expression53. Durable MTSS1 induction was achieved in CML cell lines following in vitro treatment through the demethylating agent 5-azacytidine. The apoptotic activator BCL2-like protein (BIM) has been demonstrated to undergo epigenetic reprogramming after TKI therapy and downregulated BIM levels are linked to decreased optimal reactions54. IM and the demethylating agent 5-aza-deoxycytidine together increased the expression of BIM and reduced the viability and cell proliferation of CML cell lines54. Short strands of non-coding RNAs (ncRNAs) called microRNAs (miRNAs) frequently bind to the 3’ UTR of a target mRNA, and either stop translation or cleave the mRNA55. miR-150, miR-146a and miR-10a expression were significantly lower at CML diagnosis when compared to healthy individuals; however, following a short period of IM therapy, their expression levels returned to normal56-58. The expression profiles of several microRNAs, including the miR-17-92 cluster, also known as oncomir-1, differ in CML. At the time of diagnosis, CD34+ cells from CML patients had higher expression levels of oncomir-159. Within 14 days of beginning IM therapy, Flamant et al. discovered a reduction in oncomir-1 expression in MNC58.
BCR-ABL1 dependent mechanism of resistance
BCR-ABL1 kinase domain (KD) mutation
A point mutation in the KD of BCR-ABL oncoprotein changes its configuration due to which TKIs can no longer attach to it effectively. As the chemical structure and mechanism of action of each TKI drug are slightly different, one TKI may be able to overcome the resistance from a mutation that another TKI cannot. Mutations are responsible for generating resistance in nearly two-thirds of resistant CML-AP and CML-BP patients and around one-third of resistant CML-CP patients.
T315I gatekeeper mutation, identified as the first BCR-ABL KD mutation, provides resistance against first and 2GTKIs. However, Dasatinib helped to overcome most Imatinib-resistant mutations but was not able to overcome the T315I mutation60. This mutation can be treated with drugs like Ponatinib and Synribo (Omacetaxine). Other resistant point mutations are Y253F, F317L, G250E, M351T and V256G, and two novel frameshift mutations are Glu281 and Tyr393. All these mutations affect the P-loop, gatekeeper, activation and catalytic loop domain region of BCR-ABL protein and cause poor Imatinib binding in the ATP binding region61.
There are plenty of clinical trials ongoing for treating CML under different types of mutations, drug resistance, and checking the efficacy of new or combinations of drugs. Some of the registered clinical trials (Supplementary Table) and drugs are as follows:
Drugs under clinical trial
The role of TKIs has a fixed position in first-line treatment; therefore, most of the trials focus on TFR or TKI holidays. A distinct category of clinical trials in CML is researching developing treatments behind relapse, with their main concern on Ponatinib and Bosutinib. Other encouraging trials involve a combination of TKI with various agents that target non-BCR-ABL1 proteins. Additionally, clinical trials are going on for third-line (3L) treatment where new CML therapies targeting BCR-ABL1 are in development, including drugs like PF114, HQ1351 and Asciminib.
PF114 (NCT02885766, Phase I/II)
An orally administered fourth-generation TKI, is effective at nanomolar concentration against both wild BCR-ABL1 and mutated BCR-ABL1, along with T315I mutation62. PF114 is anatomically similar to ponatinib but varies to avoid obstruction of vascular endothelial growth factor (VEGF) receptors to curb cardiovascular toxicity.
HQP1351 (NCT04126681, Phase II)
Also called Olverembatinib, is an orally delivered drug (1-60 mg) of the third-generation BCR-ABL1 TKI. Olverembatinib shows in-vitro action in opposition to T315I, other mutants and non-mutated BCR-ABL1. It has displayed remarkable and durable efficiency in phase I trial63.
ASCIMINIB (ABL001)
It is an allosteric BCR-ABL TKI which binds to the myristoyl binding pocket of the kinase domain of ABL1. ABLOO1 does not bind to the ATP binding site and therefore, maintains substantial activity against kinase domain mutation that imparts resistance to other TKIs. The safety and potency of ABLOO1 are being evaluated in phase I (NCT02081378), II (NCT03578367) and III (NCT03106779) trials and currently approved by FDA in October 2021.
Presence of BCR-ABL1 oncogene
As described in the earlier section, BCR-ABL1oncoprotein can be controlled by TKIs, while BCR-ABL1 oncogene remains unchanged, which can produce a more significant number of BCR-ABL1 transcripts generating oncoprotein, leading to CML. With the advancements in genome editing tools, many researchers are working hard to eliminate the oncogene and treat CML. This can act as a future hope to cure CML as described in the following section.
Future hope to cure CML: CRISPR/Cas
To overcome the issue of the production of more oncoproteins from unaltered BCR-ABL1 oncogene, genome editing tools can be used as new alternative therapeutics. The opportunity to knock down oncogenes is now practical with the emergence of genome-editing nucleases like transcription activator-like effector nucleases (TALENs), zinc finger nucleases (ZFNs) and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated protein (Cas). Among various genome editing tools, CRISPR/Cas is under extensive research for treating genetic disorders.
Research work on CRISPR/Cas9 in the field of leukaemia has immensely accelerated in the past few years64-68. The evolution of mouse models that imitate human CML69 has granted new possibilities to assess the therapeutic function of CRISPR/Cas9. CRISPR/Cas9 tool perhaps rectifies the acquired mutations of the human leukaemia cell line70. In 2016, the foremost clinical trials engaging CRISPR/Cas9 work in humans began71. It was reported in 2017 that the CRISPR/Cas9 system could successfully abolish the BCR-ABL1 oncogene64. Nullification of BCR-ABL1 oncogene by another genome editing nuclease, ZFNs, was demonstrated in 201865. Recently, it was reported that the human CML cell line (K562) displayed a diminishing proliferation rate after the use of the CRISPR/Cas9 lentiviral vector to obstruct ABL1 in BCR-ABL1 oncogene72, which has exposed the therapeutic capability of CRISPR/Cas system. CRISPR/Cas 9 functioned as the therapeutic system by obstructing the BCR-ABL1 oncogene by aiming at ABL1 exon 267, fusion sequence68 and ABL1 exon 673 while ZFNs obstructed the BCR exon 165.
This genome editing tool still possesses some technical limitations, but the number of available substitutes to conquer them has accelerated at the same rate. A reliable method of delivery is the significant limitation of in-vivo CRISPR/Cas therapy. The CRISPR toolkit can be delivered as Cas9 mRNA and gRNA or as plasmid DNA74. Popular viral vectors for in-vivo CRISPR component delivery include Adeno-associated-virus (AAV), Lentivirus (LV) and Adeno-virus (AdV). Despite their high in vivo transfection efficiency, concerns about clinical use remain, such as immunogenicity and integration75. Another concern is the pre-existing adaptive immunity against Cas9 in humans, necessitating the use of new Cas proteins. Off-target effects (OTEs) are common when using CRISPR/Cas9 for gene therapy74. Another limitation of the technology is the requirement for a nearby PAM sequence. Streptococcus pyogenes Cas9 (SpCas9), a commonly used variant, detects a short PAM sequence (5′NGG3′). However, its size challenges gene therapy delivery via AAV vectors. To broaden the gene target range, various SpCas9 variants have emerged, like SpCas9-NG and xCas976,77. Even though CRISPR editing in humans is still a hotly debated and contentious subject, a few FDA-approved and RAC-reviewed CRISPR gene therapy trials have begun after careful analysis of the risk-to-benefit ratios. These initial approved trials, which are now in Phase I/II, are only for patients with severe illnesses, like cancers or incapacitating monogenic diseases.
In the future, it is expected that the CRISPR/Cas9 therapy will turn out to be routine clinical practice.
Conclusions
Chronic myeloid leukaemia can be treated by chemotherapy, immunotherapy and targeted therapy, however, both chemotherapy and immunotherapy are less efficacious than targeted therapy. Targeted therapy chiefly uses TKIs to block the ATP binding pocket of BCR-ABL1 oncoprotein and therefore, inhibits kinase activity. Currently, there are four TKIs (Imatinib, Nilotinib, Dasatinib and Bosutinib) permitted to be used in first-line treatment, while these can also be used in second-line treatment. All TKIs are recommended according to the patient’s age, comorbidity, phase of the disease and drug toxicity profile. Numerous clinical trials in different phases are ongoing with distinct strategic approaches, like the combination of TKIs with chemotherapeutic agents and drugs that aim at non-BCR-ABL1 targets. Researchers are working on an alternative approach where genome editing tools can be used, like CRISPR/Cas9 and ZFNs, to eliminate the BCR-ABL1 oncogene, in the hope of curing CML. However, this method contains limitations like optimal delivery method, off-target cleavage and pre-existing adaptive immunity to Cas9. It is expected that these limitations will be resolved in the future and there will be a definite cure for CML.
Financial support & sponsorship
The work of ST was supported by the Department of Biotechnology (DBT), grant number [DBT/2021-22/JNU/1844].
The work of AS was supported by Indian Council of Medical Research (ICMR), grant number PAC-SCSM-HRK-ICMR-04220423-1440.
Conflicts of Interest
None
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of Artificial Intelligence (AI)-Assisted Technology for assisting in the writing of the manuscript and no images were manipulated using AI.
References
- Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin. 2021;71:209-49.
- [CrossRef] [Google Scholar]
- An overview and update of chronic myeloid leukemia for primary care physicians. Korean J Fam Med.. 2015;36:197-202.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The biology of chronic myeloid leukemia. N Engl J Med. 1999;341:164-72.
- [CrossRef] [PubMed] [Google Scholar]
- High-risk additional chromosomal abnormalities at low blast counts herald death by CML. Leukemia. 2020;34:2074-86.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Regional variations in age at diagnosis and overall survival among patients with chronic myeloid leukemia from low and middle income countries. Cancer Epidemiol. 2013;37:247-54.
- [CrossRef] [PubMed] [Google Scholar]
- Magnitude and temporal trend of the chronic myeloid leukemia: On the basis of the Global Burden of Disease Study 2019. JCO Glob Oncol. 2021;7:1429-41.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Comparison of chromosome banding analysis, interphase- and hypermetaphase-FISH, qualitative and quantitative PCR for diagnosis and for follow-up in chronic myeloid leukemia: A study on 350 cases. Leukemia. 2002;16:53-9.
- [CrossRef] [PubMed] [Google Scholar]
- Monitoring disease burden in chronic myeloid leukemia: Past, present, and future. Am J Hematol. 2016;91:742-6.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular testing in CML between old and new methods: Are we at a turning point? J Clin Med. 2020;9:1-16.
- [CrossRef] [Google Scholar]
- Dr Lal PathLabs. Available from: https://www.lalpathlabs.com, accessed on August 16, 2023
- DNA Labs India. Available from: https://dnalabsindia.com/, accessed on August 16, 2023.
- ClinLab Navigator. Chronic Myeloid Leukemia Tests. Available from: https://www.clinlabnavigator.com/chronic-myeloid-leukemia-tests.html, accessed on August 17, 2023.
- Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994-1004.
- [CrossRef] [PubMed] [Google Scholar]
- Final 5-year study results of DASISION: The dasatinib versus imatinib study in treatment-Naïve chronic myeloid leukemia patients trial. J Clin Oncol.. 2016;34:2333-40.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Long-term benefits and risks of frontline nilotinib vs. imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. 2016;30:1044-54.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: Results from the randomized BFORE trial. J Clin Oncol. 2018;36:231-7.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Phase III clinical trial (RERISE study) results of efficacy and safety of radotinib compared with imatinib in newly diagnosed chronic phase chronic myeloid leukemia. Clin Cancer Res. 2017;23:7180-8.
- [CrossRef] [PubMed] [Google Scholar]
- Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: Final 5-year results of the phase 2 PACE trial. Blood. 2018;132:393-404.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The effects of imatinib mesylate treatment before allogeneic transplantation for chronic myeloid leukemia. Blood. 2007;109:1782-9.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Nilotinib in patients with Ph + chronic myeloid leukemia in accelerated phase following imatinib resistance or intolerance: 24-month follow-up results. Leukemia. 2012;26:1189-94.
- [CrossRef] [PubMed] [Google Scholar]
- Standardized definitions of molecular response in chronic myeloid leukemia. Leukemia. 2012;26:2172-5.
- [CrossRef] [PubMed] [Google Scholar]
- Deep molecular response is reached by the majority of patients treated with imatinib, predicts survival, and is achieved more quickly by optimized high-dose imatinib: Results from the randomized CML-Study IV. J Clin Oncol. 2014;32:415-23.
- [CrossRef] [PubMed] [Google Scholar]
- Moving treatment-free remission into mainstream clinical practice in CML. Blood. 2016;128:17-23.
- [CrossRef] [PubMed] [Google Scholar]
- Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11:1029-35.
- [CrossRef] [PubMed] [Google Scholar]
- Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): A multicentre phase 2 trial. Lancet Haematol. 2015;2:e528-e535.
- [CrossRef] [PubMed] [Google Scholar]
- Discontinuation of dasatinib or nilotinib in chronic myeloid leukemia: Interim analysis of the STOP 2G-TKI study. Blood. 2017;129:846-54.
- [CrossRef] [PubMed] [Google Scholar]
- Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO-SKI): A prespecified interim analysis of a prospective, multicentre, non-randomised, trial. Lancet Oncol. 2018;19:747-57.
- [CrossRef] [PubMed] [Google Scholar]
- Durable treatment-free remission in patients with chronic myeloid leukemia in chronic phase following frontline nilotinib: 96-week update of the ENESTfreedom study. J Cancer Res Clin Oncol. 2018;144:945-54.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- ENESTOP 192-wk results: Durability and impact on quality of life of treatment-free remission (TFR) following second-line (2L) nilotinib (NIL) in patients (PTS) with chronic myeloid leukemia (CML) Hemasphere. 2019;2:1-19.
- [Google Scholar]
- Treatment-free remission in CML: The US perspective. Curr Hematol Malig Rep. 2019;14:56-61.
- [CrossRef] [PubMed] [Google Scholar]
- Targeted therapies: Remembrance of things past - Discontinuation of second-generation TKI therapy for CML. Nat Rev Clin Oncol. 2017;14:201-2.
- [CrossRef] [PubMed] [Google Scholar]
- Droplet digital PCR is a robust tool for monitoring minimal residual disease in adult Philadelphia-positive acute lymphoblastic leukemia. J Mol Diagnostics. 2018;20:474-82.
- [CrossRef] [Google Scholar]
- Detection and quantification of BCR-ABL1 fusion transcripts by droplet digital PCR. J Mol Diagnostics. 2014;16:174-9.
- [CrossRef] [Google Scholar]
- Quantitative assessment of the BCR-ABL transcript using the cepheid Xpert BCR-ABL Monitor assay. Arch Pathol Lab Med. 2007;131:947-50.
- [CrossRef] [PubMed] [Google Scholar]
- Cepheid xpert monitor platform for the confirmation of BCR-ABL1 IS conversion factors for the molecular monitoring of chronic myeloid leukaemia. Leuk Res. 2016;49:47-50.
- [CrossRef] [PubMed] [Google Scholar]
- Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol Cancer. 2018;17:1-15.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Molecular response to imatinib and its correlation with mRNA expression levels of imatinib influx transporter (OCT1) in Indian Chronic Myeloid Leukemia patients. Asian Pacific J Cancer Prev. 2017;18:2043-8.
- [Google Scholar]
- OCT1 and imatinib transport in CML: Is it clinically relevant? Leukemia. 2015;29:1960-9.
- [CrossRef] [PubMed] [Google Scholar]
- Drug transporters play a key role in the complex process of Imatinib resistance in vitro. Leuk Res. 2015;39:355-60.
- [CrossRef] [PubMed] [Google Scholar]
- Interaction of the Efflux transporters ABCB1 and ABCG2 with imatinib, nilotinib, and dasatinib. Clin Pharmacol Ther. 2014;95:294-306.
- [CrossRef] [PubMed] [Google Scholar]
- ABCG2 - A transporter for all seasons. FEBS Lett. 2004;567:116-20.
- [CrossRef] [PubMed] [Google Scholar]
- Resistance to tyrosine kinase inhibitors in chronic myeloid leukemia—from molecular mechanisms to clinical relevance. Cancers (Basel). 2021;13:1-36.
- [Google Scholar]
- High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood. 2011;117:3409-20.
- [CrossRef] [PubMed] [Google Scholar]
- A role for FOXO1 in BCR-ABL1-independent tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Leukemia. 2016;30:1493-501.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- A therapeutically targetable mechanism of BCR-ABL-independent imatinib resistance in chronic myeloid leukemia. Sci Transl Med. 2014;6:252ra121.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Epigenetic reprogramming sensitizes CML stem cells to combined EZH2 and tyrosine kinase inhibition. Cancer Discov. 2016;6:1248-57.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458:591-6.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell. 2012;21:266-81.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Loss of β-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12:528-41.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Genetic and pharmacologic inhibition of β-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell. 2012;10:412-24.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Abrogation of histone deacetylases (HDACs) decreases survival of chronic myeloid leukemia cells: New insight into attenuating effects of the PI3K/c-Myc axis on panobinostat cytotoxicity. Cell Biol Int. 2021;45:1111-21.
- [CrossRef] [PubMed] [Google Scholar]
- Mtss1 is a critical epigenetically regulated tumor suppressor in CML. Leukemia. 2016;30:823-32.
- [CrossRef] [PubMed] [Google Scholar]
- Epigenetic down-regulation of BIM expression is associated with reduced optimal responses to imatinib treatment in chronic myeloid leukaemia. Eur J Cancer. 2009;45:1877-89.
- [CrossRef] [PubMed] [Google Scholar]
- MicroRNAs: Small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522-31.
- [CrossRef] [PubMed] [Google Scholar]
- Down-regulation of hsa-miR-10a in chronic myeloid leukemia CD34+ cells increases USF2-mediated cell growth. Mol Cancer Res. 2008;6:1830-40.
- [CrossRef] [PubMed] [Google Scholar]
- Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol Cancer. 2011;10:1-13.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Micro-RNA response to imatinib mesylate in patients with chronic myeloid leukemia. Haematologica. 2010;95:1325-33.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Expression of the miR-17-92 polycistron in chronic myeloid leukemia (CML) CD34+ cells. Blood. 2007;109:4399-405.
- [CrossRef] [PubMed] [Google Scholar]
- The structure of dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 2006;66:5790-7.
- [CrossRef] [PubMed] [Google Scholar]
- Novel mutations in the kinase domain of BCR-ABL gene causing imatinib resistance in chronic myeloid leukemia patients. Sci Rep. 2019;9:1-17.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- PF-114: A 4th generation tyrosine kinase-inhibitor for chronic phase chronic myeloid leukaemia including BCRABL1T315I. Blood. 2019;134:1638.
- [CrossRef] [Google Scholar]
- An updated safety and efficacy results of phase 1 study of HQP1351, a novel 3rd generation of BCR-ABL tyrosine kinase inhibitor (TKI), in patients with TKI resistant chronic myeloid leukemia. Blood. 2019;134:493.
- [CrossRef] [PubMed] [Google Scholar]
- The CRISPR/Cas9 system efficiently reverts the tumorigenic ability of BCR/ABL in vitro and in a xenograft model of chronic myeloid leukemia. Oncotarget. 2017;8:26027-40.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Induction of apoptosis in imatinib sensitive and resistant chronic myeloid leukemia cells by efficient disruption of BCR-ABL oncogene with zinc finger nucleases. J Exp Clin Cancer Res. 2018;37:1-14.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Splice donor site sgRNAs enhance CRISPR/ Cas9-mediated knockout efficiency. PLoS One. 2019;14:1-19.
- [Google Scholar]
- Efficient disruption of BCR-ABL gene by CRISPR RNA-guided FokI nucleases depresses the oncogenesis of chronic myeloid leukemia cells. J Exp Clin Cancer Res. 2019;38:1-13.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- In vivo CRISPR/Cas9 targeting of fusion oncogenes for selective elimination of cancer cells. Nat Commun. 2020;11:5060.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Chronic myeloid leukemia (CML) mouse model in translational research. Methods Mol Biol. 2016;1438:225-43.
- [CrossRef] [PubMed] [Google Scholar]
- ASXL1 mutation correction by CRISPR/Cas9 restores gene function in leukemia cells and increases survival in mouse xenografts. Oncotarget. 2015;6:44061-71.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- CRISPR gene-editing tested in a person for the first time. Nature. 2016;539:479.
- [CrossRef] [PubMed] [Google Scholar]
- ABL genomic editing sufficiently abolishes oncogenesis of human chronic myeloid leukemia cells in vitro and in vivo. Cancers (Basel). 2020;12:1-25.
- [CrossRef] [Google Scholar]
- CRISPR / Cas9 technology abolishes the BCR/ABL1 oncogene in chronic myeloid leukemia and restores normal hematopoiesis. bioRxiv 2020.08.05.237610; doi: https://doi.org/10.1101/2020.08.05.237610; 2020; 4 : 1037290
- [PubMed] [Google Scholar]
- CRISPR gene therapy: Applications, limitations, and implications for the future. Front Oncol. 2020;10:1387.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Viral vectors for the in vivo delivery of CRISPR components: Advances and challenges. Front Bioeng Biotechnol. 2022;10:1-6.
- [CrossRef] [Google Scholar]
- Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259-62.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556:57-63.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




